
1. Introduction.
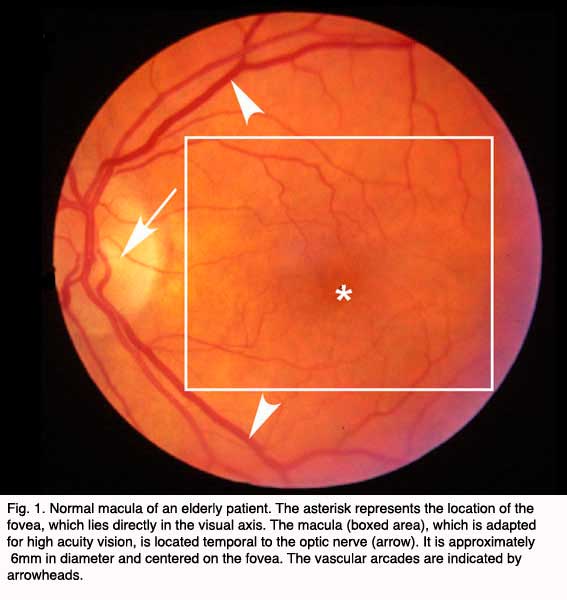
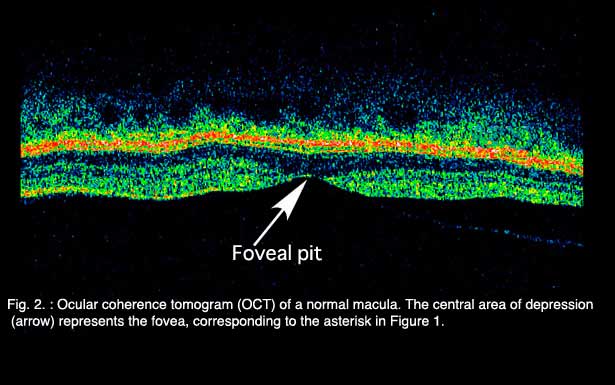
Age-related macular degeneration (AMD), the leading cause of worldwide blindness in the elderly, is a bilateral ocular condition that affects the central area of retina known as the macula. The macula lutea, which derives its name from the deposition of yellow xanthophyll pigments (see chapter on Simple Anatomy), is located temporal to the optic disc and is bounded by the temporal superior and inferior vascular arcades (Fig. 1). Although the macula comprises only four percent of retinal area, it is responsible for the majority of useful photopic vision. The fovea lies at the center of the macula (Fig. 1, asterisk) and is approximately 2mm in diameter. The fovea is particularly well seen in vertical section view using ocular coherence tomography techniques in living eyes (Fig. 2). The fovea contains the highest density of cone photoreceptor cells and is the only region of the retina where 20/20 vision is attainable. The macula accounts for almost 10% of the entire visual field. Thus, lesions developing in this region can have a major impact on visual function.
 |  |
AMD has a tremendous impact on the physical and mental health of the geriatric population and their families. Prior to 1990, AMD of all forms was often referred to as “senile macular degeneration” or SMD, a reflection of the fact that the vision loss associated with AMD manifests late in life when most affected individuals are looking forward to enjoying retirement activities and maintaining independence. Instead, millions with AMD suffer bilateral central vision loss such that they can no longer drive, read a newspaper, prepare meals, or enjoy recreational activities. For many patients, the visual impairment associated with AMD means a loss of independence, depression, increased financial concerns and the need to adapt to vision loss at a time when they are likely suffering from other debilitating conditions(1-5).
The cost to society is only now being appreciated. A recent analysis of AMD in Australia predicts that the disease costs $2.6 billion per year (6). This is projected to grow to $6.5 billion by 2025, a total cost of $59 billion over the next 20 years. A treatment that reduced the progression by only 10% would save Australia $5.7 billion over that same period of time. Similar analyses for the United States are lacking, but given the demographics and higher cost of medical care in the US, the costs would be projected to as much as twenty-fold higher.
Clinically, AMD is classified into the nonexudative “dry” or atrophic form and the exudative “wet” or neovascular form. More severe vision loss is typically associated with the “wet” form that occurs in about 15% of all patients with AMD but up to 20% of legal blindness from AMD is due to the “dry” form (7).
2. Clinical Aspects of AMD.
Clinical Presentation of AMD
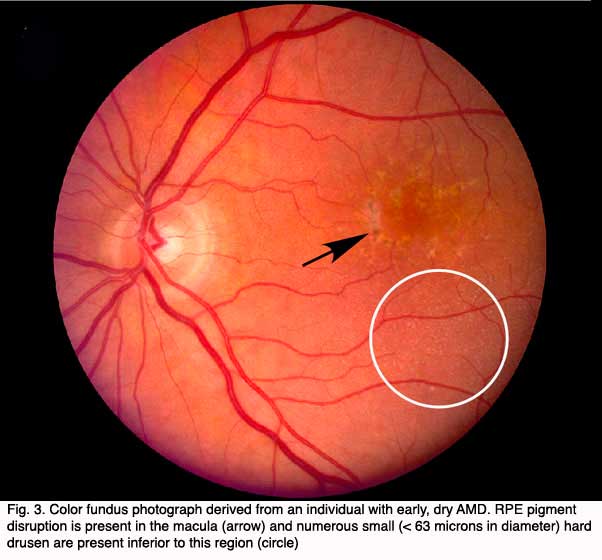
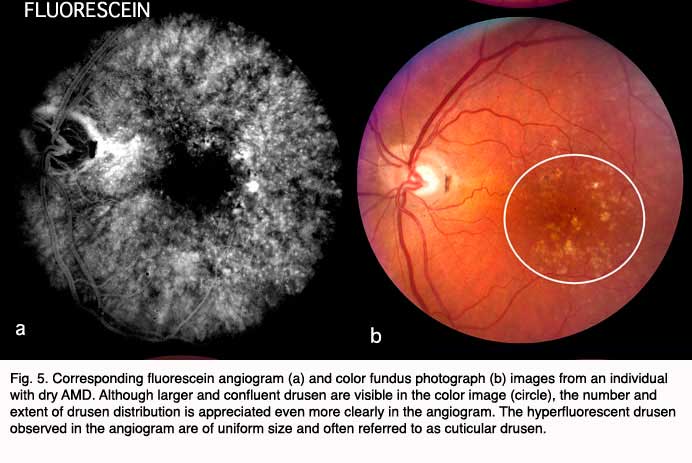
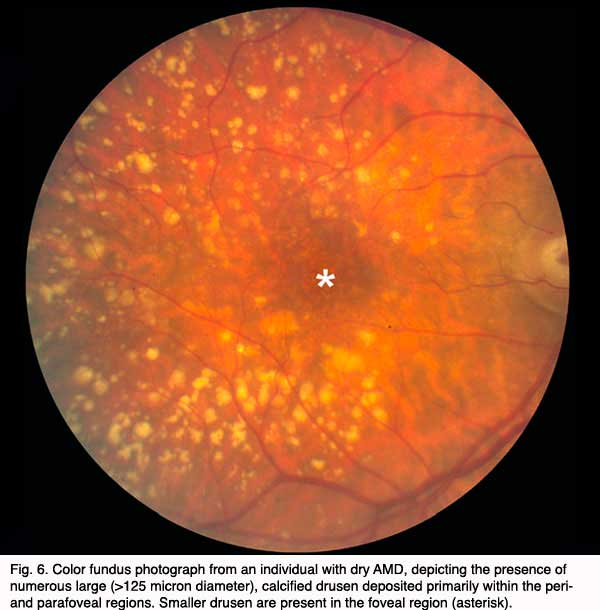
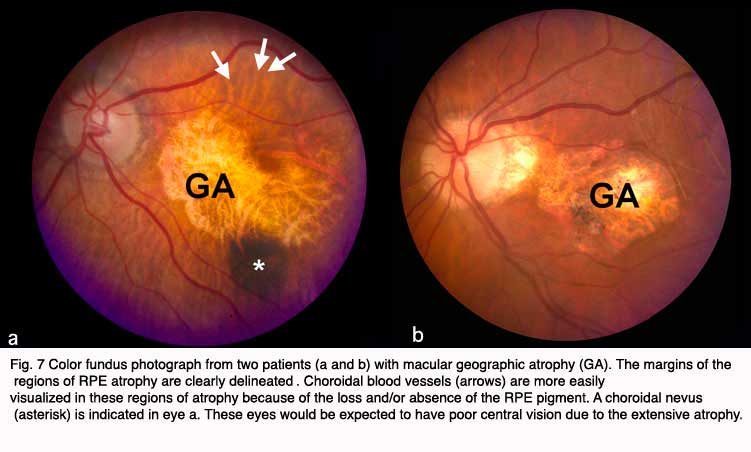
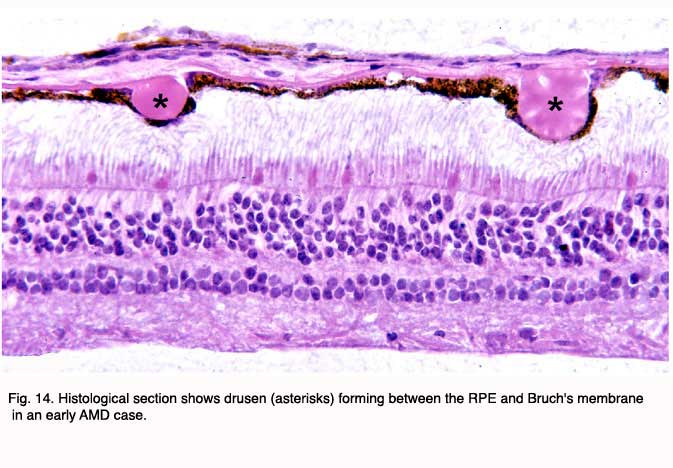
Although autopsy studies have documented histological and ultrastructural changes associated with AMD in the retina, retinal pigment epithelium (RPE), choriorcapillaris and choroid in middle age, the disease typically does not manifest clinically before age 55 (10). AMD is generally thought to progress along a continuum from atrophic or “dry” AMD (Figs. 3 and 4) to neovascular “wet” AMD with approximately 10% – 15% of all AMD patients eventually developing the wet form. Occasionally patients with no prior signs of dry AMD present with exudative changes as the first manifestation of the condition. The typical clinical sign of “dry” AMD is pigment disruption and drusen (small yellowish deposits in Figs. 3, 4 and 5) in the retina. Drusen may be small “hard” (small with discrete margins) or “soft” (larger with indistinct edges) (Figs. 3, 4 and 5). They lie between the RPE and an adjacent basement membrane complex known as Bruch’s membrane (BM) (See later Figs. 13-16). Geographic atrophy (GA) and RPE changes are also observed in dry AMD (Fig. 7). Several classification schemes have been developed that subdivide dry AMD into categories based on the number and size of drusen, amount of GA, and degree of pigmentary changes in the macula (25-28).
 |  |
Angiography using sodium fluorescein dye is commonly performed when signs of AMD are observed and and particularly when exudative changes are suspected.
The natural history of dry AMD is progressive, with gradual loss of visual function that may span over many years time. Many patients with dry AMD are asymptomatic and unaware of the disease. In 10-15% percent of patients with dry AMD the deterioration is more rapid and extensive and they suffer significant vision loss due to geographic atrophy.
In approximately 10-15% of patients the condition progresses to the “wet” or neovascular form. Left untreated, the natural history of wet AMD advances further to a cicatrical stage referred to as a disciform scar. This process usually takes place over several months and typically results in a 4-8 mm diameter fibrotic scar underlying the macula accompanied by a central scotoma with severe central vision loss in one eye (Fig. 11). Patients with neovascular AMD in one eye have a 4 – 12% per year cumulative risk of developing neovascular AMD in the fellow eye. Thus, the risk of bilateral loss of central vision is high in those with the neovascular form of the disease (30).
The designation of exudative or “wet” AMD implies that fluid, exudates and/or blood are present in the extracellular space between the neural retina and the RPE (i.e. the subretinal space) and/or, as in the case of RPE detachments, between the RPE and Bruch’s membrane (i.e. the sub-RPE space).
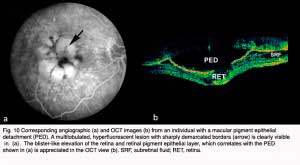
The neovascular tissue associated with exudative AMD is most commonly referred to as choroidal neovascularization (CNV) because it originates from the normal choriorcapillaris and extends through a dehiscence in Bruch’s membrane into the subretinal or sub-RPE space. Sometimes multiple soft drusen become confluent and create large pigment epithelial detachments (PED’s), which represent an elevation of the retinal pigment epithelium under the retina (Fig.10). Neovascularization associated with PEDs can be difficult to visualize clinically or image using fluorescein angiography; therefore, it is difficult to treat (29).
The following figures show pictures of different patients’ retinas with advanced cases of AMD of the wet form. Figure 8 shows a color fundus picture (a) and an early (b) and late fluorescein angiogram (c) from an individual with a classic choroidal neovascularization (CNV). Subretinal blood and fluid (arrowhead depicts edge) is clearly visible within the macular region in (a). The blood vessels have a lacey appearance (asterisk) in the early angiogram. Marked leakage of fluorescein (asterisk), with indistinct edges of hyperfluorescence (white area) is clearly visible in the late angiogram.
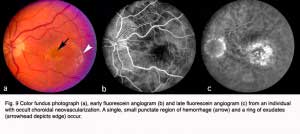
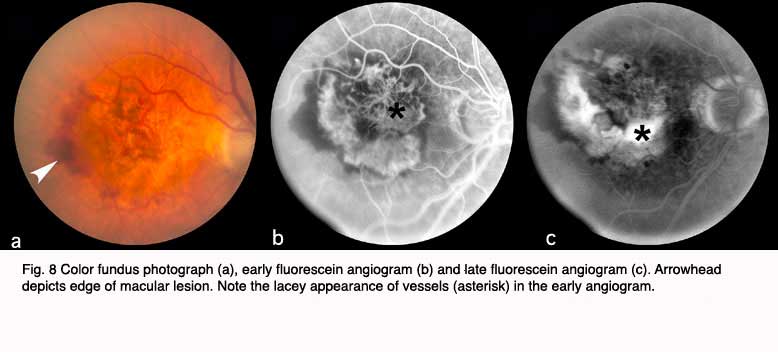 Figure 9 shows a similar set of fundus pictures from an individual with occult choroidal neovascularization. A single, small punctate region of hemorrhage (arrow) and a ring of exudates (arrowhead depicts edge) appear more pronounced than typically occur in classic CNV. This patients had a minimal hyperfluorescence in the early phase of the angiogram, in contrast to that observed in the early stage of classic CNV. Speckled macular hyperfluorescence is visible in the late stage angiogram (Fig. 9c).
Figure 9 shows a similar set of fundus pictures from an individual with occult choroidal neovascularization. A single, small punctate region of hemorrhage (arrow) and a ring of exudates (arrowhead depicts edge) appear more pronounced than typically occur in classic CNV. This patients had a minimal hyperfluorescence in the early phase of the angiogram, in contrast to that observed in the early stage of classic CNV. Speckled macular hyperfluorescence is visible in the late stage angiogram (Fig. 9c).
 In the case shown in Figure10, the individual had a macular pigment epithelial detachment (PED). A multilobulated, hyperfluorescent lesion with sharply demarcated borders (arrow) is clearly visible in the late stage angiogram of Figure 10a, arrow. The OCT shown in Figure 10b, contrasts with a normal OCT of the fovea (Fig. 2) by being a blister-like elevation of the retina and retinal pigment epithelial layer off Bruch’s membrane and the choroid.
In the case shown in Figure10, the individual had a macular pigment epithelial detachment (PED). A multilobulated, hyperfluorescent lesion with sharply demarcated borders (arrow) is clearly visible in the late stage angiogram of Figure 10a, arrow. The OCT shown in Figure 10b, contrasts with a normal OCT of the fovea (Fig. 2) by being a blister-like elevation of the retina and retinal pigment epithelial layer off Bruch’s membrane and the choroid.
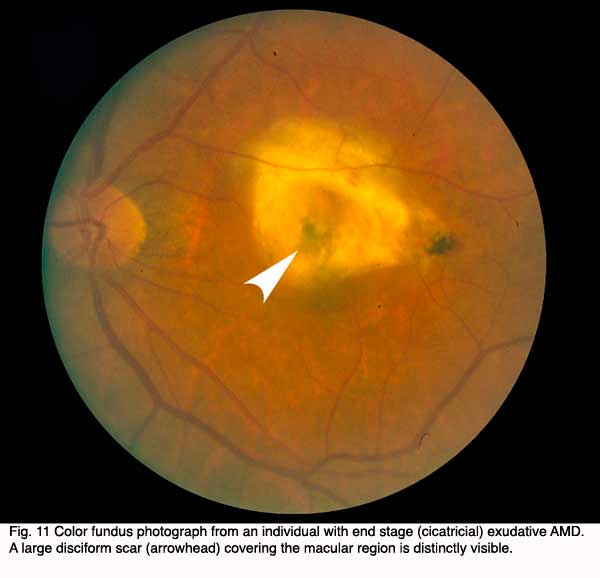
 The end stage of such lesions of the macula as shown in the three previous patients is illustrated by the last patient (Fig. 11). The color fundus photograph shows that the patient has end stage (cicatricial) exudative AMD. A large disciform scar (arrowhead) covers the whole macular region. Such a patient would be almost totally blind in that eye.
The end stage of such lesions of the macula as shown in the three previous patients is illustrated by the last patient (Fig. 11). The color fundus photograph shows that the patient has end stage (cicatricial) exudative AMD. A large disciform scar (arrowhead) covers the whole macular region. Such a patient would be almost totally blind in that eye.
3. Pathology of AMD.
Overview
Until quite recently, the majority of research on AMD and the development of therapeutics for the disease has focused upon late-stage neovascular events. No clear-cut sequence for the initiation and progression of AMD has been identified that would allow it to be monitored and treated in its earliest stages. Moreover, the pathologic correlates of the different clinical phenotypes of AMD remain poorly understood, and there is a paucity of animal models that accurately mimic the characteristic features of the disease. Nevertheless, recent discoveries are beginning to provide a much clearer picture of the relevant cellular events, genetic factors, and biochemical processes that are associated with early AMD. This new information should hasten significantly the development of clinically effective diagnostics and therapeutics for the treatment of this devastating condition.
Morphological Correlates of Early AMD
From a histopathologic standpoint, the earliest detectable changes associated with AMD occur at the interface between the macular retina and the underlying layer of connective tissue and blood vessels known as the choroid (66-70). At this location lie the outer segments of rod and cone photoreceptors, the retinal pigment epithelial (RPE) cells, a stratified basement membrane complex termed Bruch’s membrane (BM), and the choroidal capillary bed or choriocapillaris (Figs. 12, 13, BV of choroid).

The initial clinical diagnosis of early AMD is based on seeing drusen – the hallmark indicators of disease – and/or pigmentary changes in the macula. Drusen look like yellow-white spots in the retina (Figures 3-7). They are extracellular deposits located between the retinal pigment epithelial (RPE) basal lamina and the inner collagenous layer of the elastin-containing Bruch’s membrane as illustrated in the schematic of Fig. 13 (71); The normal retinal choroidal interface is shown in comparison to the drusen bearing abnormal interface. Fig 14 shows an actual histological section of retina where drusen are seen between the choroid and Bruch’s membrane.
 |  |
Bruch’s membrane consists of an elastin core flanked on both sides by a collagenous layer and a basal lamina (see Fig. 13) (76-79). It is widely believed that the barrier properties of the RPE and Bruch’s membrane limit cellular migration, especially the invasion of neovascular tissue from the choroid into the subretinal space. Our understanding of the molecular composition of Bruch’s membrane, the molecular traffic that occurs across it, and the homeostatic mechanisms that maintain it, during normal aging as well as in AMD, is still rudimentary (80).
Links between AMD and structural abnormalities in Bruch’s membrane have been documented in numerous histopathologic studies. Classically, these Bruch’s membrane defects have been described as fragmentation or fracturing in association with calcification (81-85). Additional age-related changes in Bruch’s membrane are typified by; a) Progressive thickening of the two collagenous layers; b) Modification and degeneration of collagen and elastin; c) Increased levels of advanced glycation end products, noncollagenous proteins and lipids; and d) Accumulation of several types of sub-RPE deposits (Fig. 13)(86-95). It has also been suggested that the age-related abnormalities in Bruch’s membrane eventually lead to photoreceptor degeneration as a result of increased hydrophobicity, reduced permeability, and impaired nutrient exchange between the choroid and the RPE. Functionally, such changes result in an exponential reduction in the hydraulic conductivity of Bruch’s membrane as a function of age (96-99). Much less is known about changes in Bruch’s membrane hydraulic conductivity in individuals with AMD.
It has been proposed that topographic variations in Bruch’s membrane may render the macula more susceptible to the ingrowth of new blood vessels from the choroidal vasculature that characterizes neovascular AMD. Morphometric data indicate that the elastic lamina of Bruch’s membrane in the macula is 3-6 fold thinner and 2-5 fold less dense relative to that in the mid-periphery in individuals of all ages (100). Elastin fiber destruction in the macula could also play a key role in the initiation of neovascular events because elastin degradation peptides are highly angiogenic and possess macrophage recruiting activity (101, 102).
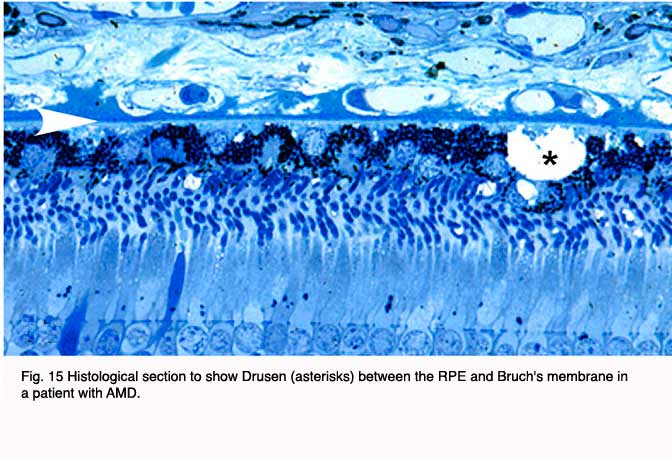 | 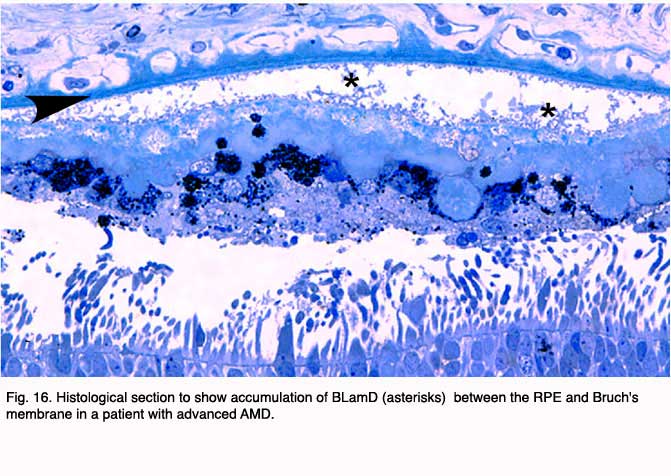 |
In addition to drusen, basal laminar deposits (BLamD) and basal linear deposits (BLinD) accumulate within the extracellular environment of the RPE-choroid interface (Figs. 13, 15, 16 and 17). These are identified based upon their morphological appearance and location. Both types of deposits have been proposed by various investigators to be involved in the pathogenesis of AMD (103). Neutral lipid (including esterified cholesterol) and apolipoproteins B and E are abundant in these basal deposits and in drusen in the eyes of aged donors and donors with a documented history of AMD (88, 94, 104-107).

Basal laminar deposits (BLamD) that accumulate between the RPE basal plasma membrane and its basal lamina (Figs. 13, 15, 16 and 17) do not appear to be specific to or ‘diagnostic’ for AMD. However, there is a strong relationship between macular BLamD and exudative AMD, disciform scarring, and visual loss (74, 84, 108-115). BLamD-like deposits are also seen in other macular disorders with a clinical appearance similar to what is observed in AMD. These include Sorsby’s fundus dystrophy, Late Onset Retinal Degeneration (LORD), adult foveomacular pigment epithelial dystrophy and Malattia leventinese (116). The molecular composition and origin of BLamD has not yet been identified (117-119), but its morphological similarity to long-spacing collagen is striking. Electron microscopy has revealed that its banding pattern matches that of type VI collagen (120).
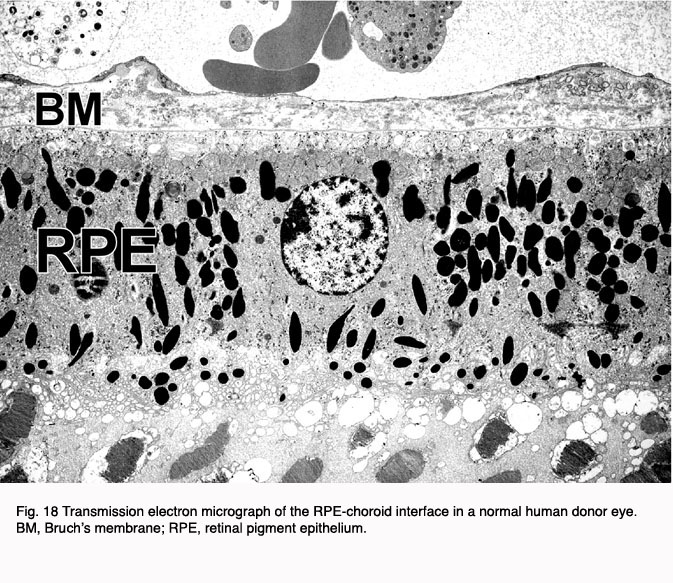
Electron microscopy shows the changes that occur at the choroids RPB interface in AMD at a more detailed level. Figures 18 and 19 indicate a normal retinal choroidal interface (Fig. 18) compared with an abnormal AMD-like situation where there are drusen forming at this interface (Fig. 19).

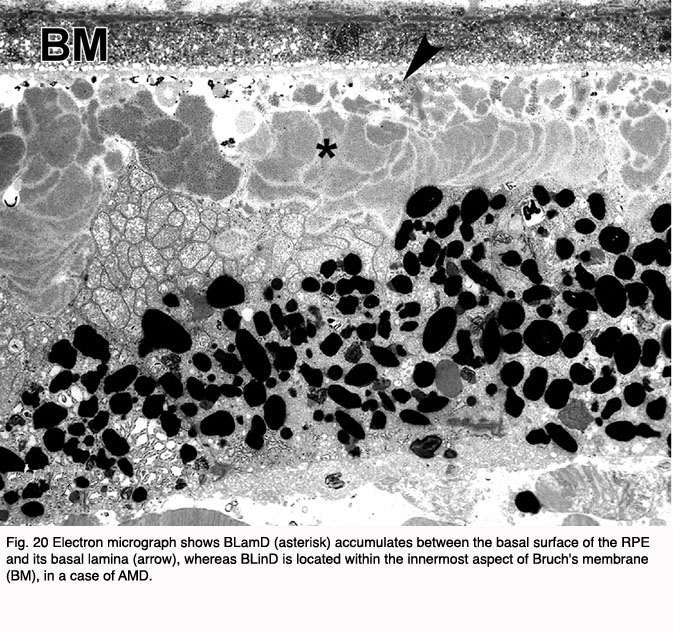
BlinD lies between the RPE basal lamina and the elastic layer of Bruch’s membrane (Figure 20). The principal component of BLinD is a distinct form of membranous debris, thought to be comprised primarily of lipoprotein particles containing neutral lipids, including esterified cholesterol. It is likely that BLinD is derived from local ocular sources (e.g. RPE cells) (121). BLinD were initially described by Sarks and colleagues (122) as lipid-like vesicles with varying diameters.
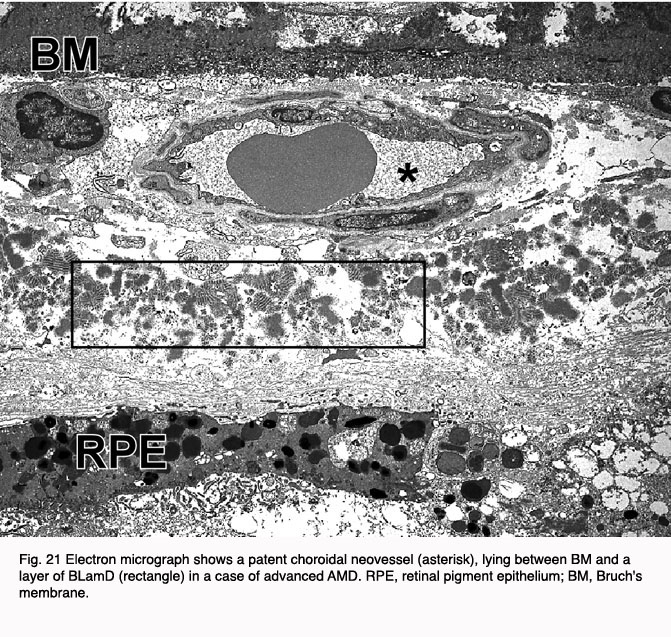
It is believed that the pigmentary changes often observed in the macula of AMD eyes are attributable to degenerative changes in the highly melanized RPE cells (Fig. 20). This notion has gained wide acceptance because most of the early clinical signs and histopathological changes have been localized to this cell layer (123-126). It has long been recognized that the RPE is essential for the transport of ions, nutrients, and metabolites from the circulation to the neural retina and vice versa (127). If the RPE interface to the Bruch’s membrane is broken down, amongst other things, blood vessels from the choroids can grow into this space (Fig. 21).
A viable RPE is also essential for normal photoreceptor cell metabolism and functioning of the visual cycle. Therefore, it is not surprising that RPE degeneration is accompanied by concomitant photoreceptor degeneration. RPE dysfunction in the macula, whether caused by local environmental insults and/or genetic defect(s), would have a profound impact on the overlying photoreceptors and, therefore, on central vision. However, the nature of the insult(s) that causes RPE dysfunction has yet to be ascertained, although a variety of hypotheses have been advanced over the years including genetic factors, ischemia, oxidative stress, phagocytic overload, cigarette smoke, lipofuscin toxicity and, most recently, microbial infection (128-134).
The choriocapillaris has also been suspected to play a role in the etiology of AMD. The density and diameters of the choriocapillaris capillaries decreases with age, and this decrease is even greater in patients with AMD. The overall loss appears most marked in regions of geographic atrophy (135, 136).
4. Physiology of early AMD.
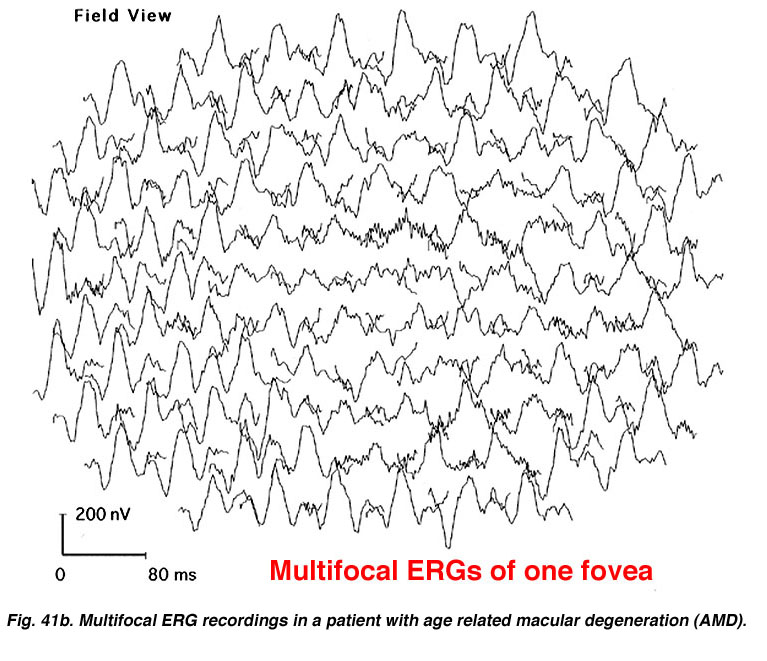
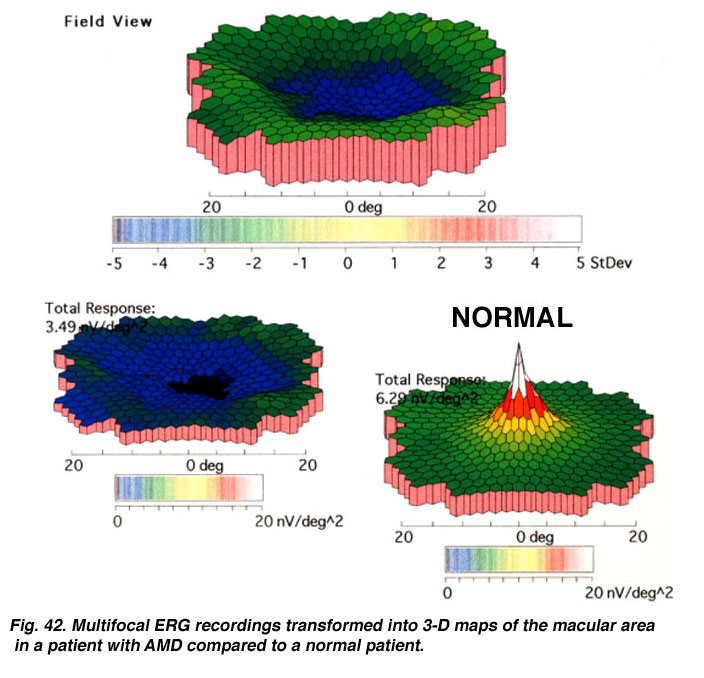
There is substantial evidence of photoreceptor involvement in early AMD, particularly in those patients with soft or large drusen in the macula (137-145). Contrast sensitivity, the rate of recovery after photostress, the amplitude and latency of the foveal electroretinogram (ERG) response, and dark adaptation are all affected adversely in early AMD patients, including those with normal visual acuity (140, 146-151). Figure 22 Shows the multifocal ERG responses from the macular area of a patient with AMD (See chapter on the Clinical ERG, multifocal ERG). The responses of the fovea are reduced in amplitude. In the 3-D map (Fig. 23) it can be seen that the foveal area is flat, suggesting no cone activity, compared with the characteristic peak of responses in the normal retina.
It has been shown that photoreceptor cells in areas impacted by drusen exhibit morphologic (FIGURE??) and biochemical signs of degeneration, including decreased expression of synapse-associated proteins and increased expression of stress-response molecules. There are also drusen-associated reductions in photoreceptor cell densities, thus suggesting that degenerative changes in photoreceptors ultimately lead to cell death (152, 153).
5. Inflammation and AMD.
Recent studies of the molecular composition of drusen have implicated local inflammation as a key element in the pathogenesis of AMD (154-160). Drusen contain numerous proteins related to the process of inflammation or its aftermath (106, 161-164). In particular, many of these proteins are associated with the complement cascade and its regulation. Some of the proteinaceous components in drusen and the sub-RPE space are activated complement components and fragments associated with assembly of the membrane attack complex (MAC) (162, 165-168). Other drusen components including vitronectin (161), clusterin, complement receptor 1 (CR1), and membrane cofactor protein (MCP-1) (164) are known complement regulatory proteins. Still others include known activators of the complement cascade such as cholesterol (169), C-reactive protein (155), and the amyloidogenic peptide amyloid b (153, 170). Metallic zinc, an amyloid b binding molecule, is also a drusen constituent (171). This compositional profile forms the basis for the conclusion that drusen are a manifestation of chronic, local inflammation at the level of Bruch’s membrane.
6. The Complement System and AMD.
Three complement pathways can be distinguished by the “triggers” that activate them. 1) antigen-antibody binding triggers the “classical pathway”. 2) carbohydrate residues bound to invading pathogens trigger the “lectin pathway. 3) A low level of spontaneous activation known as “tickover” characterizes the “alternative pathway”. This continuous background level of activation is amplified rapidly by ubiquitous components present on the surfaces of many bacteria, viruses, as well as constituents present in cigarette smoke.
Complement Factor H, the Complement Alternative Pathway and AMD
Factor H is the main soluble inhibitor of the “alternative pathway” and, like most complement components, the liver is responsible for 80-90% of Factor H protein synthesis (190-192). We now know that variants in the gene encoding Factor H play a central role in one’s susceptibility to AMD (see below). All of the available data are consistent with the conclusion that uncontrolled activation of the alternative pathway of complement at the level of Bruch’s membrane is a key element in the process of drusen formation and a major contributing factor to the pathogenesis of AMD.
Although the liver is responsible for most circulating Factor H, the eye is also capable of producing it, in addition to other complement components. Not surprisingly, Factor H protein is also a molecular constituent of drusen (193). It co-localizes with its ligand C3b (a complement pathway component) in substructural spherules within drusen that contain amyloid b, further implicating these structures as candidate complement activators (170, 194). Factor H and the MAC co-distribute in drusen and at the interface between the RPE and choroid. Finally, Factor H and C5b-9 (MAC) immunolabeling are more intense in the macula compared to tissues from more peripheral areas in the same eye (Hageman and Mullins, unpublished).
We have advanced a working model of AMD pathobiology based upon our studies (155, 195) and the work of others (157, 196). In this model, RPE atrophy and the subsequent deposition of cellular debris in the sub-RPE space is construed as a local pro-inflammatory “seeding” event, leading to activation of the innate immune system at the RPE-choroid interface. Complement attack, in turn, induces significant bystander damage to macular cells and tissues, thus rendering them susceptible to additional RPE atrophy, photoreceptor degeneration, and CNV.(The reader, who is interested, can see diagrams showing the way the complement cascade works, in a paper by Ricklin and Lambris, 2007) (247).
7. Molecular genetics of AMD.
Overview
AMD is often regarded as a group of complex, late onset diseases caused by the convergence of multiple risk factors (197, 198). Family history is a consistent risk factor according to most epidemiological studies of AMD conducted to date. Familial aggregation studies have shown that a genetic contribution is identified in up to 25% of AMD cases (199). Twin studies also support a genetic basis for the disease, with the concordance of clinical features (drusen and pigmentary changes) for both early and late onset disease being approximately twice as high in monozygotic (identical) twins compared to dizygotic (non-identical) twins (200, 201).
Genome-wide linkage analyses of extended families with AMD have identified a number of chromosomal loci that are linked to AMD (202-210). The most consistent occurs at chromosome 1q31 (211). Between 15-40% of multiplex AMD families segregate with a disease gene in the 1q31 region (205). Further analysis of this region reveals an allelic variant in exon 104 of the fibulin-6 gene (FBLN-6) in members of one AMD family, as well as in some sporadic AMD cases (212). A number of other candidate genes have also been linked to AMD. However, most of these results have not yet been replicated in subsequent studies. These genes include ELOVL4 (213), VEGF, VLDLR, and LRP6 (214), FIBLN5 (215) and TLR-4 (216).
In contrast, the e4 allele of the apolipoprotein E (APOE) gene has consistently been shown to be protective for AMD (217-219), whereas the e2 allele appears to influence progression and to result in an earlier age of onset (219). ABCA4 confers increased risk for AMD too (220).
In early 2005, four groups reported independently that common variants [single nucleotide polymorphisms (SNPs)] in the gene encoding complement Factor H (CFH) confer major susceptibility to, or protection from, AMD (193, 221-223). Individuals who possess a single copy of the risk-conferring CFH haplotype have 2-4 fold higher lifetime risk of developing AMD, and those with two copies possess a 5-7 fold higher lifetime risk. Haplotype analysis showed that a SNP in the CFH gene can account for up to 50% of AMD cases in the human population, a number approaching 50,000,000 worldwide. Follow-up reports have since confirmed these findings in additional cohorts in the United States (213, 224), Iceland (225), the United Kingdom (226), and France (227). Interestingly, the major CFH risk haplotype in the Japanese population appears to be different than that reported in other populations, although the protective haplotype is the same (228).
In early 2006, a study showed that allelic variants in two other complement-related genes, Factor B (BF) and complement component C2, were linked to AMD (229). These are paralogous genes located 500bp apart on chromosome 6p21, and both reside in the major histocompatibility complex (MHC) class III region. BF is a component of the alternative pathway of complement, whereas C2 is a component of the classical pathway. Both variants together can account for nearly 74% of the risk of developing AMD. The protective haplotype for each of the three genes was completely absent in nearly 75% of all AMD cases; whereas 56% of controls possessed at least one copy of a protective CFH or BF haplotype. Approximately 60% of the risk in AMD cases, and 65% of the protection in controls, can be assigned to the CFH gene locus alone.
In addition to the 1q31 locus, a region on chromosome 10q26 has been identified in several recent studies of AMD (211) on a scale similar to that of the CFH Tyr402His variant. PLEKHA1 (pleckstrin homology domain-containing, family A, member 1) and a predicted LOC387715 gene were identified as the relevant candidate genes at that locus (231). A subsequent study concluded that SNP rs10490924 in LOC387715 was the most likely AMD-susceptibility allele (232). A third study identified a coding change in the LOC387715 gene as the significant allele that results in a non-synonymous substitution of alanine to serine at position 69 of the hypothetical protein. In addition, a significant statistical relationship between the LOC387715 variant and a history of cigarette smoking has been found (233). Thus far, there is no published evidence for expression of the LOC387715 gene product at either the RNA or protein levels; nor is there any indication of its likely functional properties.
8. Therapy for AMD.
The molecular pathogenesis of AMD has only recently begun to be elucidated. So, it is not surprising that the therapeutic approaches for AMD have been of only limited benefit to most patients. Therapeutic interventions have focused almost exclusively upon the exudative “wet” form that comprises approximately 10% of the AMD patient population because it is the most debilitating and rapidly progressive form of the disease. Until recently, such therapies have resulted in a benefit to less than 50% of patients with “wet” AMD, and therefore less than 5% of all AMD patients. Developing therapeutics to delay onset or progression of “dry” AMD or the conversion from “dry” to “wet” AMD, have proven even more difficult. This due to the long study duration and large numbers of participants needed to achieve meaningful statistical results.
Over the last 30 years epidemiological studies have repeatedly identified four risk factors for AMD: age, cigarette smoking, increased body mass index, and inheritance (10, 31, 32). These same studies have produced conflicting information regarding the roles of dietary antioxidant supplementation and fat intake. In 1992 the Age-related Eye Disease Study (AREDS) group began enrolling subjects in a prospective, multicenter, randomized, controlled study(33). Patients enrolled in the AREDS had either mild, moderate, or severe dry AMD in both eyes, or exudative AMD in one eye only. The study tried to determine whether antioxidant therapy prevented vision loss, slowed the progression of dry AMD, or prevented conversion from dry to wet AMD. The results of 6.3 years of follow-up in the AREDS study were published in 2001 (33). The study results demonstrated that daily antioxidant therapy in the form of 15 mg beta carotene, 500 mg vitamin C, 50 mg vitamin E, 80 mg zinc (as zinc oxide), and 2mg copper (as cupric oxide) daily was superior to a placebo in delaying progression of advanced dry AMD, and in slowing conversion from dry to wet AMD. Specifically, the odds ratio of progression to advanced AMD was decreased to 0.72 in the group who took the above combination of antioxidants as compared to placebo. No benefit of antioxidant supplementation was demonstrated in the milder forms of AMD. However, because the early stages of AMD often persist for many years, it is quite possible that the study design was not long enough to demonstrate a benefit to patients with milder stages of AMD.
Another therapeutic intervention is laser therapy for eyes with drusen. Three prospective trials, the Choroidal Neovascularization Prevention Trial (CNVPT), Complications of AMD Prevention Trial (CAPT) and Prophylactic Treatment of Age-Related Macular Degeneration Trial (PTAMD) evaluated the efficacy and safety of light macular grid laser in reducing the risk of developing CNV in eyes with drusen. The CNVPT and CAPT used argon laser and the PTAMD used an 810 nm diode laser (34-36). In both the CNVPT and PTAMD studies, grid laser treatment increased the risk of CNV. Therefore grid laser of eyes with drusen is not recommended if the fellow eye has CNV (36, 37). Recent results of the PTAMD bilateral drusen study reported no decrease in CNV development but a four-letter gain in visual acuity in treated versus control eyes after three years of follow-up (38). The larger CAPT study of treatment of one eye in the setting of bilateral drusen is ongoing but near completion of five years of follow-up. Until the results of the CAPT study are known, prophylactic laser of eyes with bilateral drusen is not a recommended therapy.
Therapy for exudative AMD has evolved from thermal laser photocoagulation in the early to mid 1980’s, to photodynamic therapy for CNV beginning in 2000 and, most recently to inhibition of vascular growth factors. The three year results of the Macular Photocoagulation Studies (MPS) for AMD was published in 1986. It demonstrated a benefit for “extrafoveal” CNV located between 200 and 3,000 microns from the fovea (Fig. 24). Subsequent MPS studies for juxtafoveal (1 – 200 microns from the fovea) CNV and subfoveal CNV respectively, demonstrated a benefit for laser treatment versus no treatment (Fig. 24). (39-41).
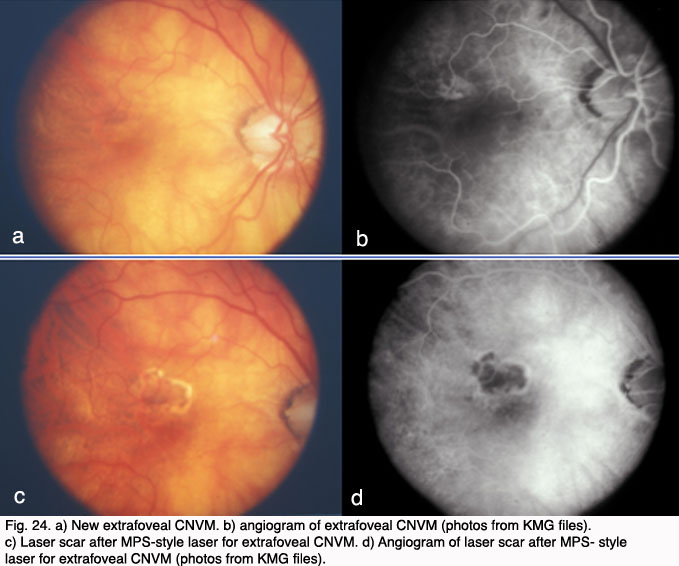
However, visual loss was common even in those eyes with successful obliteration of CNV (42). Additionally, the closer the CNV was to the fovea, the poorer the visual outcome and the higher the rate of recurrence even in those who initially responded to treatment (43). In 1993, Freund and coworkers demonstrated that only 13% of new patients with exudative AMD (or approximately 1.3% of all AMD patients) met criteria for thermal laser treatment based on the MPS (44).
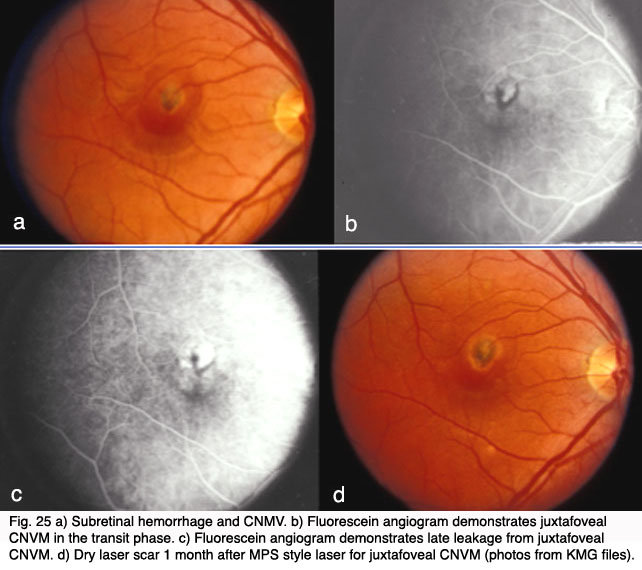
In the late 1980’s to mid-1990’s there was significant interest in vitreoretinal surgical approaches to exudative AMD. Vitrectomy surgery with evacuation of subretinal blood and neovascular membranes reported variable success rates. Trial designs were nonstandard, results were conflicting, and long-term follow-up in these pilot trials was lacking. Given the potential morbidity and expense of major eye surgery, the National Eye Institute sponsored the prospective, randomized, controlled Submacular Surgery Trials (SST). The results of the trial demonstrated that surgery did not increase the chance of stable or improved vision, and it prevented severe vision loss only in those eyes with large subretinal hemorrhages. Submacular surgery was also associated with some risk of rhegmatogenous retinal detachment. Given these results, and the advent of less-invasive therapy with anti VEGF agents (described below) submacular surgery is no longer recommended for exudative AMD except, perhaps, in select cases with large submacular hemorrhage(45, 46).
Other surgical approaches, which have been investigated in small pilot trials, are macular translocation (Machemer and Steinhorst, 47), and limited macular translocation developed later by deJuan (48). The former technique involves creation of a total retinal detachment, while the latter is limited to a partial retinal detachment. Both techniques involve displacement of the macular retina from an area of CNV to a location where the retinal pigment epithelium is thought to be healthier. While pilot trials have suggested a benefit from these therapies for a small subset of patients, the emergence of less invasive and more broadly applicable pharmacological therapies for exudative AMD have resulted in much less frequent application of macular translocation in recent years.
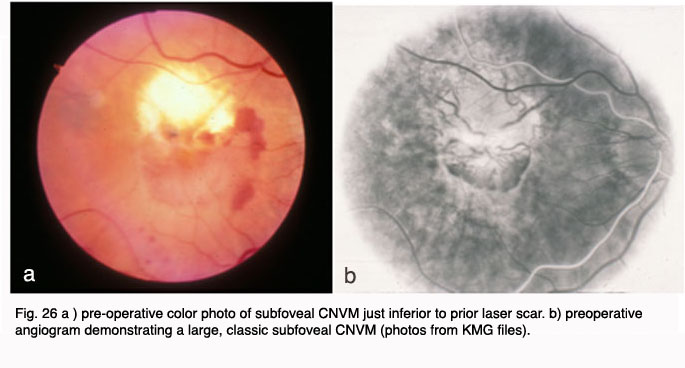
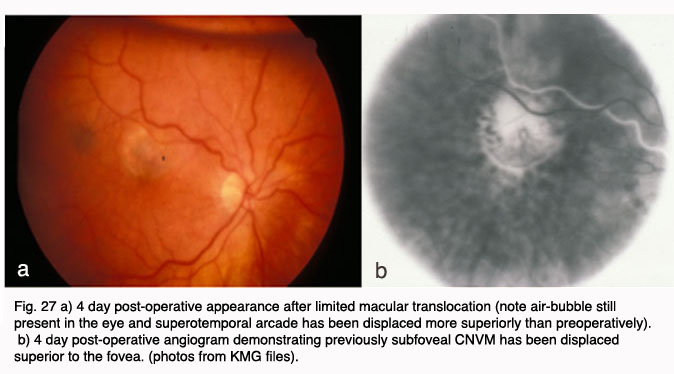
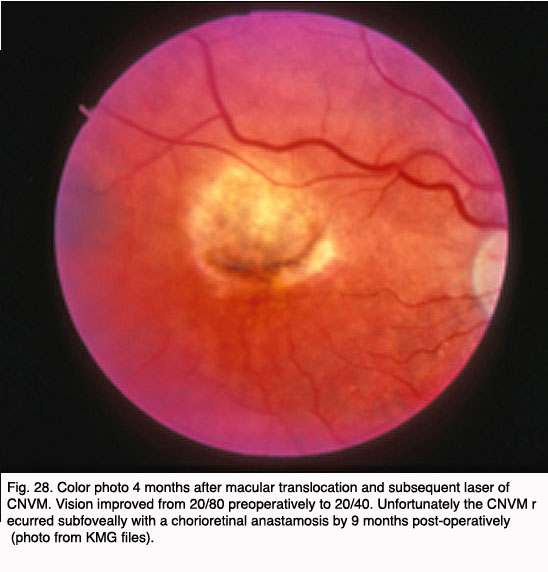
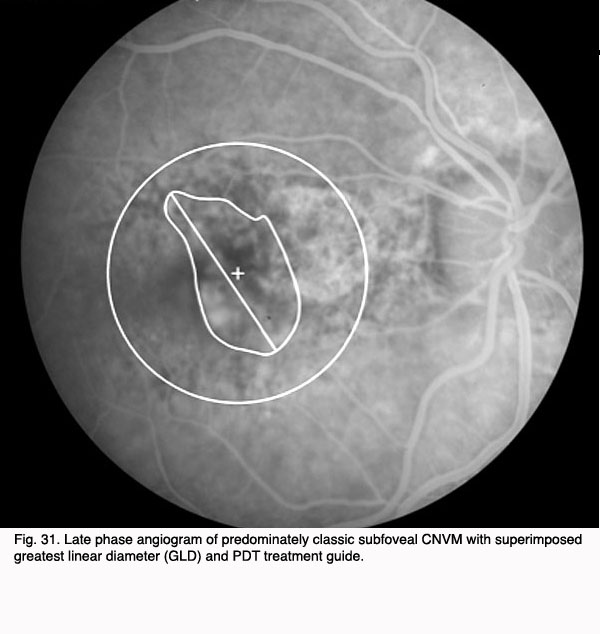
In 2000, photodynamic therapy with Visudyne ™ (verteporfin), a light-activated compound, was approved by the FDA for exudative AMD with predominately (>50%) classic, subfoveal CNV that was no larger than 5600 microns in diameter. FDA approval was based on the results of the Treatment of Age-Related Macular Degeneration with Photodynamic Therapy Investigation (TAP) study. In the TAP study, subfoveal CNV was divided into subgroups based on initial visual acuity, lesion size, and fluorescein leakage characteristics: enrolled subjects were randomized to Visudyne(tm) or placebo (49). Photodynamic therapy (PDT) involves intravenous administration of the photoactivatable compound, Visudyne ™ followed 15 minutes later by irradiation of the CNV with a low energy, non-thermal laser. Activated Visudyne(tm) generates singlet oxygen that damages the CNV endothelium resulting in thrombosis (50). In the TAP study an average of 3.5 treatments spaced at three month intervals were required to ablate the CNV, so the process is a lengthy one. Visual results, though better than with any prior treatment, were less than optimal with only 67% of treated patients with predominately classic CNV losing fewer than three lines of visual acuity (as compared to 39% of placebo treated eyes). At 1 year, 16% of Visudyne treated patients gained one or more line of vision, and 15% of treated patients lost more than six lines of visual acuity. Over the course of the next several years, subgroup analysis of data from the TAP studies as well as additional trials with Visudyne(tm) treatment for lesions other than predominately classic CNV led to its application for other lesion types. However, the visual results for lesions that were predominantly occult CNV were less favorable than those with predominately classic CNV (51, 52).
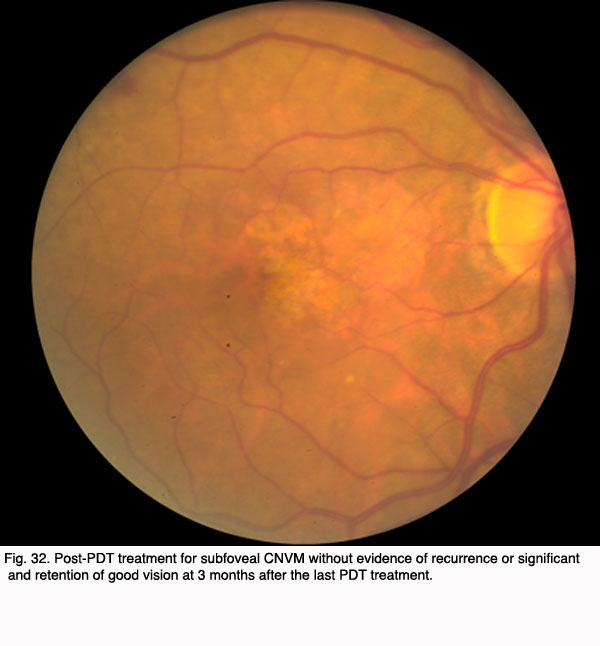
The Figures 29 to 32 demonstrate a favorable response to PDT with Visudyne as seen in our clinic.
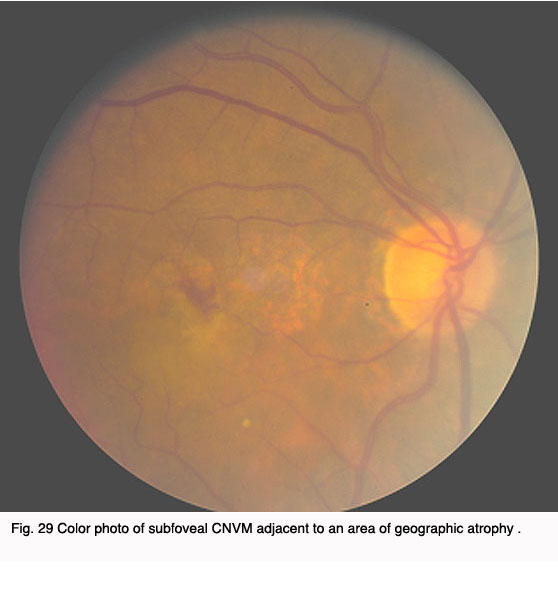 Fig. 29 Color photo of subfoveal CNVM adjacent to an area of geographic atrophy |  Fig. 30. Angiogram demonstrating predominately classic subfoveal CNVM adjacent to an area of geographic atrophy |
As noted above, however, not all eyes fare well with PDT therapy. Below is a photograph of an eye that did not demonstrate a significant benefit from PDT treatment with Visudyne.
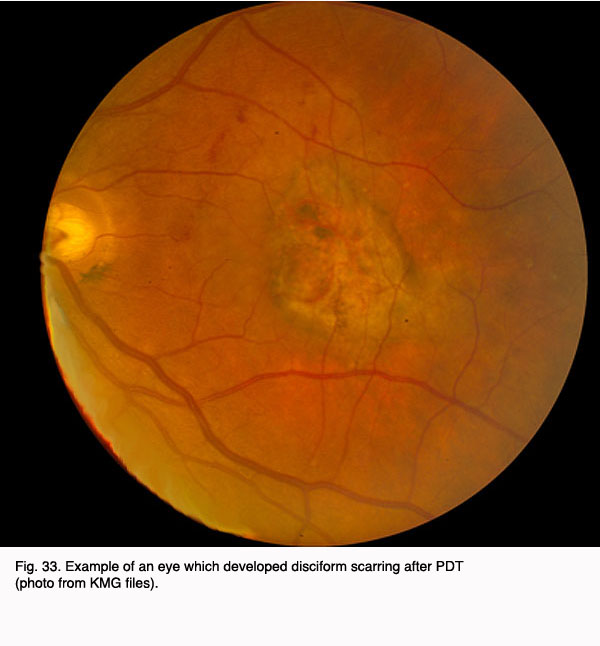
Fig. 33. Example of an eye which developed disciform scarring after PDT (photo from KMG files)
Other ablative treatments for CNV include transpupillary thermotherapy (TTT) and radiation therapy. TTT utilizes low energy diode laser (810 nm) applied to the CNV slowly over one minute. Small pilot trials of TTT for occult CNV initially showed promise but a randomized, prospective clinical trial (TTT4 CNV) demonstrated no significant benefit. Similarly, low dose radiation therapy for AMD was investigated in multiple small pilot trials with conflicting results (53-55). Although no definitive prospective clinical trial of radiation therapy for AMD has ever been conducted, the concept has become less popular in recent years, especially in light of the interest in pharmacological therapies for AMD.
The current era of therapy for exudative AMD utilizes administration of anti-neovascular agents periocularly or intraocularly. Studies implicating various cellular growth factors, including vascular endothelial growth factor (VEGF) in a variety of ocular neovascular processes has led to the development of VEGF inhibitors, VEGF antibodies and other “broad-spectrum” antiangiogenic molecules that inhibit pro-angiogenic cytokines. In 2004, Macugen(tm) (Pegantanib Sodium) was the first of the VEGF inhibitors to be approved by the FDA for exudative AMD. Macugen is an aptamer which binds to the A isoform of VEGF-165 at its heparin binding site and prevents VEGF-165 from binding to its receptor on endothelial cells. Macugen is administered by intravitreal injection (i.e. injected into the vitreous cavity of the eye) every 6 weeks for up to a two-year course of therapy. Based on the Macugen ™ study results of 1186 patients after one-year, 70% of subjects treated with 0.3 mg pegaptanib, 70% lost fewer than 3 lines of vision compared to 55% of controls. The risk of severe vision loss (> 6 lines of vision) was reduced from 22% in controls to 10% in treated subjects (56).
Another VEGF inhibitor undergoing clinical trials is ranibizumab or Lucentis(tm). Lucentis(tm) is derived from the Fab fragment of an antibody to isoform A of VEGF. The fragment blocks VEGF binding to its receptor on endothelial cells, thus inhibiting its biological activity. The ANCHOR trial was presented recently (57). The data demonstrated not only a preservation of vision in nearly 95% of patients treated with Lucentis(tm) (ranibizumab) at year one but, for the first time, an improvement in vision in 30-38% of patients with exudative AMD. Another recently published trial comparing Lucentis alone to PDT alone demonstrated superior visual results with Lucentis. (ref: Anchor Trial). Lucentis(tm) was FDA approved for the treatment of exudative AMD in June 2006. Although the initial trials with Lucentis utilized monthly intravitreal injection with the drug, more recent studies indicate that some patients my respond favorably to a treatment regimen utilizing fewer than 24 injections over a 2 year period (Fung et al., 2007) (248). Currently, studies are underway to determine factors which might influence frequency and duration of treatment with Lucentis.
Avastin(tm) (beclumizabab), a VEGF antibody derived from the same parent antibody as Lucentis(tm), is approved by the FDA to treat colon cancer and is gaining popularity as an “off-label” intravitreal therapy for AMD. It is important to note that Avastin(tm) has not yet undergone the toxicity studies and controlled trials that are required by the FDA for ocular application (58, 59). Other anti-angiogenic therapies currently under study include anecortave acetate (Retane, an antiangiogenic steroid (60, 61)), squalene and triamcinolone (which carries a significant risk of causing cataract and steroid-induced glaucoma). Additional studies are underway to investigate the potential for a combination of Anti-VEGF therapy with Visudyne(tm) photodynamic therapy utilizing light doses lower than those applied in the TAP an VIP studies with Visudyne(62, 63, 249).
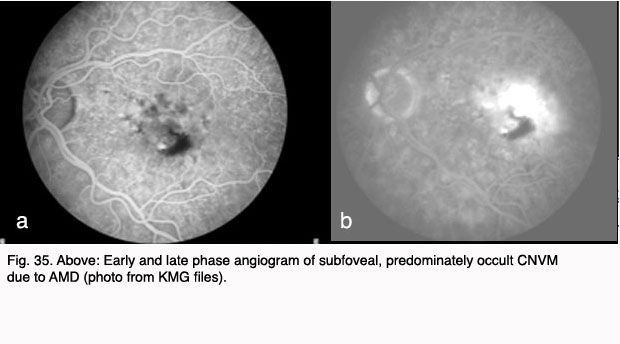
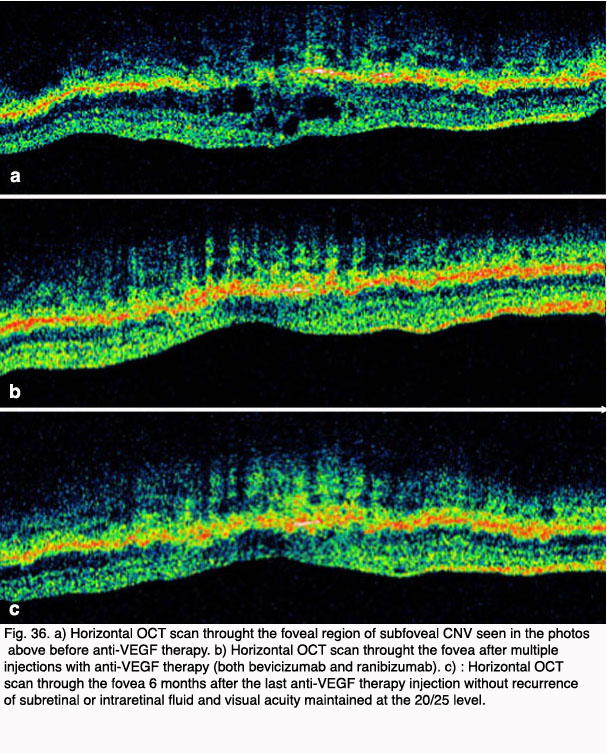
Figures 34 and 35 show results of pre (Fig. 34, 35) and post anti-VEGF treatment (Fig. 36).
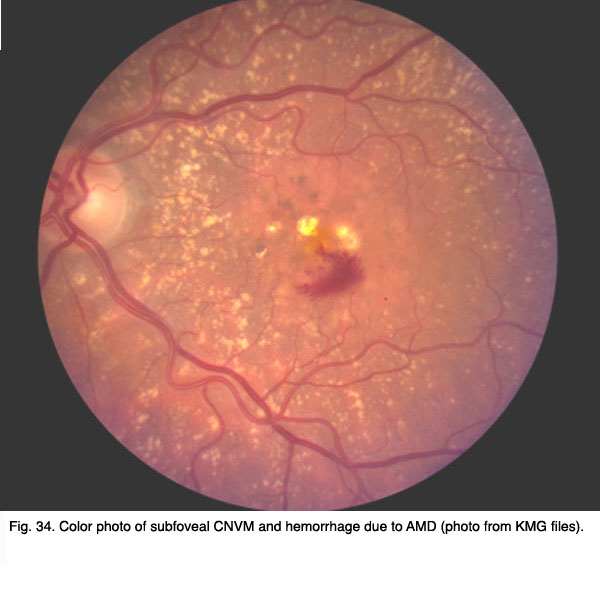
Fig. 34. Color photo of subfoveal CNVM and hemorrhage due to AMD (photo from KMG files)
{kind=link}
{kind=link}
{kind=link}
As a result of the anti-VEGF treatment much improvement can be seen in the macular area along with good visual restoration. The OCT scans (Fig. 36) show filling in of the disrupted cell types in the retina (the holes of Fig 36a get occupied by cells) and glial cells react to restore damaged retina (Figs 36b and c, green upward streaks).
One concern with all anti-angiogenic agents is their potential for systemic inhibition of VEGF that may produce cardiovascular or cerebrovascular complications, or inhibit wound healing. The immediate and long-term systemic effects of these drugs, especially those prescribed “off-label”, are largely unknown and difficult to determine in an elderly population already prone to vascular events. Additional drawbacks to the intravitreally administered therapeutics include patient discomfort, intraocular infection (endophthalmitis), retinal tear or detachment, and lens damage in phakic patients (64). The incidence of endophthalmitis in published trials with Macugen and Lucentis were 0.16%/injection and 1.3% – 1.4% of patients respectively and the incidence of retinal tears and/or detachment in these studies were 0.08%/injection and 0% – 0.4% respectively. (see Macugen and Lucentis trials refs below) Risk of lens damage in phakic patients treated with intravitreal anti-VEGF therapy can be minimized by visualization of the needle tip with the indirect ophthalmoscope during injection and by proper patient preparation (anesthetic, education, and reassurance) during the injection process to minimize the risk of sudden patient movement. Some patients do quite well with topical anesthetic alone while others require subconjunctival anesthetic injection (249,250,251, 252).
Recent discoveries of the role that inflammation plays in AMD pathogenesis (see above) has led to an interest in investigating the anti-inflammatory effects of statins on AMD. Several clinical population-based studies of the effects of statins on AMD development and progression have yielded conflicting results (65). Thus, more studies in this area will be required to assess efficacy.
In the near future, therapy for exudative AMD will likely involve multiple anti-neovascular agents, possibly coupled with less frequent application of photodynamic therapy, in order to maximize the beneficial effects of each and minimize the frequency of adverse side effects. Further on the horizon are anti-angiogenic agents in longer acting forms that require less frequent administration. The ultimate therapy for AMD, however, will lie in preclinical identification of those who are genetically “at risk” for the disease, coupled with preventative strategies that are designed to minimize or compensate for one’s genetic predisposition.
9. Conclusion.
Complement Factor H is the first gene identified in multiple independent studies that confers a significant risk for the development of AMD. This finding, together with the subsequent identification of AMD-associated variants in the related complement genes BF and C2, provide compelling evidence that the innate immune system and, more specifically, uncontrolled regulation of the alternative pathway of complement, plays a central role in the pathobiology of AMD. At this point, the most likely scenario is that exposure to infection or some other triggering event in genetically susceptible individuals, coupled with impaired complement regulatory function leads to the sustained activation of complement cascade, drusen formation and, eventually, development of AMD.
The emergence of this new paradigm of AMD pathogenesis sets the stage for the rapid development of early diagnostics, novel bioassays, and new animal models that faithfully mimic aspects of the disease process. Furthermore, the way is now paved for development of novel therapeutic interventions aimed at modulating the alternative pathway of complement in “at risk” individuals prior to the onset of choroidal neovascularization or geographic atrophy.
Now that the genetic basis for a major proportion of AMD cases has been elucidated, what additional scientific progress may be anticipated, and what will be the significance of these new findings for the diagnosis and treatment of AMD? In the near term, a more comprehensive understanding of the genetic basis of AMD should rapidly emerge. It will not be surprising, for example, if genetic polymorphisms in additional complement components, complement regulators and, possibly, immune system effectors and inflammatory mediators are implicated in AMD. Secondly, it may be anticipated that the gene variants linked to AMD also contribute to other prevalent age-related diseases where chronic, local inflammatory processes are involved. For example, a significant statistical relationship between the CFH Tyr402His “risk” variant and the incidence of myocardial infarction has recently been reported (243, 245, 246). Based upon this new genetic information, it will be possible to devise genetic screening tests that will identify those individuals who are most at risk of developing AMD later in life. This will enable clinicians to monitor susceptible individuals from an early age, and to develop and test new preventive treatments in the early stages of the disease. Finally, the development of new diagnostic and pharmacological approaches will be hastened by the identification of the alternative pathway of complement as a prime therapeutic target. As our understanding of the pathogenesis of AMD continues to improve, so does the prospects for new diagnostic and therapeutic approaches. Hopefully, we will eventually eradicate AMD and dramatically improve the quality of life in our older generation.
10. References.
- 1. Uhlmann RF, Larson EB, Koepsell TD, Rees TS, Duckert LG. Visual impairment and cognitive dysfunction in Alzheimer’s disease. J Gen Intern Med.1991;6(2):126–32. [PubMed]
- 2.Lin MY, Gutierrez PR, Stone KL, Yaffe K, Ensrud KE, Fink HA. et al. Vision impairment and combined vision and hearing impairment predict cognitive and functional decline in older women. J Am Geriatr Soc. 2004;52(12):1996–2002. [PubMed]
- 3.Rovner BW, Casten RJ, Tasman WS. Effect of depression on vision function in age-related macular degeneration. Arch Ophthalmol. 2002;120(8):1041–4.[PubMed]
- 4.Abyad A. In-office screening for age-related hearing and vision loss. Geriatrics. 1997 Jun;52(6):45-6, 51-4, 7.
- 5.Berman K, Brodaty H. Psychosocial effects of age-related macular degeneration. Int Psychogeriatr. 2006:1–14.
- 6.Taylor H, Guymer R, Keeffe J. The Impact of Age-Related Macular Degeneration. In: Limited AEP, editor. Melbourne: University of Melbourne; 2006. p. 1-72.
- 7.Sunness JS. The natural history of geographic atrophy, the advanced atrophic form of age-related macular degeneration. Mol Vis. 1999;5:25. [PubMed]
- 8.. Magnitude and causes of visual impairment. 2004.
- 9.Resnikoff S, Pascolini D, Etya’ale D, Kocur I, Pararajasegaram R, Pokharel GP. et al. Global data on visual impairment in the year 2002. Bull World Health Organ.2004;82(11):844–51. [PubMed]
- 10.Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. Am J Ophthalmol. 2004;137(3):486–95. [PubMed]
- 11.Smith W, Assink J, Klein R, Mitchell P, Klaver C, Klein B. et al. Risk factors for age-related macular degeneration: Pooled findings from three continents.Ophthalmology. 2001;108:697–704. [PubMed]
- 12.Mitchell P, Smith W, Attebo K, Wang J. Prevalence of age-related maculopathy in Australia. The Blue Mountains Eye Study. Ophthalmology. 1995;102:1450–60.[PubMed]
- 13.VanNewkirk M, Nanjan M, Wang J, Mitchell P, Taylor H, McCarty C. The prevalence of age-related maculopathy: the visual impairment project. Ophthalmology.2000;107:1593–600. [PubMed]
- 14.Wolfs RC, Borger PH, Ramrattan RS, Klaver CC, Hulsman CA, Hofman A. et al. Changing views on open-angle glaucoma: definitions and prevalences–The Rotterdam Study. Invest Ophthalmol Vis Sci. 2000;41(11):3309–21. [PubMed]
- 15.Mitchell P, Wang JJ, Foran S, Smith W. Five-year incidence of age-related maculopathy lesions: the Blue Mountains Eye Study. Ophthalmology.2002;109(6):1092–7. [PubMed]
- 16.Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT. et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122(4):564–72. [PubMed]
- 17.Taylor HR, Keeffe JE, Vu HT, Wang JJ, Rochtchina E, Pezzullo ML. et al. Vision loss in Australia. Med J Aust. 2005;182(11):565–8. [PubMed]
- 18.Jonasson F, Arnarsson A, Peto T, Sasaki H, Sasaki K, Bird AC. 5-year incidence of age-related maculopathy in the Reykjavik Eye Study. Ophthalmology.2005;112(1):132–8. [PubMed]
- 19.Andersen N. Age-related macular degeneration among the Inuit in Greenland. Int J Circumpolar Health. 2004;63 Suppl 2:320–3. [PubMed]
- 20.Vingerling JR, Dielemans I, Hofman A, Grobbee DE, Hijmering M, Kramer CF. et al. The prevalence of age-related maculopathy in the Rotterdam Study.Ophthalmology. 1995;102(2):205–10. [PubMed]
- 21.Krishnaiah S, Das T, Nirmalan PK, Nutheti R, Shamanna BR, Rao GN. et al. Risk factors for age-related macular degeneration: findings from the Andhra Pradesh eye disease study in South India. Invest Ophthalmol Vis Sci. 2005;46(12):4442–9. [PubMed]
- 22.Munoz B, Klein R, Rodriguez J, Snyder R, West SK. Prevalence of age-related macular degeneration in a population-based sample of Hispanic people in Arizona: Proyecto VER. Arch Ophthalmol. 2005;123(11):1575–80. [PubMed]
- 23.Leske MC, Wu SY, Hennis A, Nemesure B, Yang L, Hyman L. et al. Nine-year incidence of age-related macular degeneration in the Barbados Eye Studies.Ophthalmology. 2006;113(1):29–35. [PubMed]
- 24.Wong TY, Loon SC, Saw SM. The epidemiology of age related eye diseases in Asia. Br J Ophthalmol. 2006;90(4):506–11. [PubMed]
- 25.Klein R, Davis MD, Magli YL, Segal P, Klein BE, Hubbard L. The Wisconsin age-related maculopathy grading system. Ophthalmology. 1991;98(7):1128–34.[PubMed]
- 26.The Age-Related Eye Disease Study (AREDS); design implications. AREDS report no. 1. Control Clin Trials. 1999;20(6):573–600. [PubMed]
- 27.Davis MD, Gangnon RE, Lee LY, Hubbard LD, Klein BE, Klein R. et al. The Age-Related Eye Disease Study severity scale for age-related macular degeneration: AREDS Report No. 17. Arch Ophthalmol. 2005;123(11):1484–98. [PubMed]
- 28.Ferris FL, Davis MD, Clemons TE, Lee LY, Chew EY, Lindblad AS. et al. A simplified severity scale for age-related macular degeneration: AREDS Report No. 18.Arch Ophthalmol. 2005;123(11):1570–4. [PubMed]
- 29.Wray S, Kuwabara T, Sanderson P. Menkes’ kinky hair disease: a light and electron microscopic study of the eye. Investigative Ophthalmology. 1976;15:128–38.[PubMed]
- 30.Abugreen S, Muldrew KA, Stevenson MR, VanLeeuwen R, DeJong PT, Chakravarthy U. CNV subtype in first eyes predicts severity of ARM in fellow eyes. Br J Ophthalmol. 2003;87(3):307–11. [PubMed]
- 31.Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL. Risk factors for the incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology. 2005;112(4):533–9. [PubMed]
- 32.Thornton J, Edwards R, Mitchell P, Harrison RA, Buchan I, Kelly SP. Smoking and age-related macular degeneration: a review of association. Eye.2005;19(9):935–44. [PubMed]
- 33.A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417–36. [PubMed]
- 34.Choroidal neovascularization in the Choroidal Neovascularization Prevention Trial. The Choroidal Neovascularization Prevention Trial Research Group.Ophthalmology. 1998;105(8):1364–72. [PubMed]
- 35.The Complications of Age-Related Macular Degeneration Prevention Trial (CAPT): rationale, design and methodology. Clin Trials. 2004;1(1):91–107. [PubMed]
- 36.Friberg TR, Musch DC, Lim JI, Morse L, Freeman W, Sinclair S. Prophylactic treatment of age-related macular degeneration report number 1: 810-nanometer laser to eyes with drusen. Unilaterally eligible patients. Ophthalmology. 2006 Apr;113(4):622 e1.
- 37.Prenner JL, Rosenblatt BJ, Tolentino MJ, Ying GS, Javornik NB, Maguire MG. et al. Risk factors for choroidal neovascularization and vision loss in the fellow eye study of CNVPT. Retina. 2003;23(3):307–14. [PubMed]
- 38.Friberg T. The Prophylactic Treatment of AMD Multi-Centered Trial (PTAMD): Results From the Bilateral Study Arm. 2006.
- 39.Argon laser photocoagulation for neovascular maculopathy. Three-year results from randomized clinical trials. Macular Photocoagulation Study Group. Arch Ophthalmol. 1986;104(5):694–701. [PubMed]
- 40.Krypton laser photocoagulation for neovascular lesions of age-related macular degeneration. Results of a randomized clinical trial. Macular Photocoagulation Study Group. Arch Ophthalmol. 1990;108(6):816–24. [PubMed]
- 41.Laser photocoagulation of subfoveal neovascular lesions in age-related macular degeneration. Results of a randomized clinical trial. Macular Photocoagulation Study Group. Arch Ophthalmol. 1991;109(9):1220–31. [PubMed]
- 42.Han DP, Folk JC, Bratton AR. Visual loss after successful photocoagulation of choroidal neovascularization. Ophthalmology. 1988;95(10):1380–4. [PubMed]
- 43.Persistent and recurrent neovascularization after krypton laser photocoagulation for neovascular lesions of age-related macular degeneration. Macular Photocoagulation Study Group. Arch Ophthalmol. 1990;108(6):825–31. [PubMed]
- 44.Freund KB, Yannuzzi LA, Sorenson JA. Age-related macular degeneration and choroidal neovascularization. Am J Ophthalmol. 1993;115(6):786–91. [PubMed]
- 45.Bressler NM, Bressler SB, Childs AL, Haller JA, Hawkins BS, Lewis H. et al. Surgery for hemorrhagic choroidal neovascular lesions of age-related macular degeneration: ophthalmic findings: SST report no. 13. Ophthalmology. 2004;111(11):1993–2006. [PubMed]
- 46.Hawkins BS, Bressler NM, Miskala PH, Bressler SB, Holekamp NM, Marsh MJ. et al. Surgery for subfoveal choroidal neovascularization in age-related macular degeneration: ophthalmic findings: SST report no. 11. Ophthalmology. 2004;111(11):1967–80. [PubMed]
- 47.Machemer R, Steinhorst UH. Retinal separation, retinotomy, and macular relocation: II. A surgical approach for age-related macular degeneration? Graefes Arch Clin Exp Ophthalmol. 1993;231(11):635–41. [PubMed]
- 48.de Juan E Jr, Loewenstein A, Bressler NM, Alexander J. Translocation of the retina for management of subfoveal choroidal neovascularization II: a preliminary report in humans. Am J Ophthalmol. 1998;125(5):635–46. [PubMed]
- 49.Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: one-year results of 2 randomized clinical trials–TAP report. Treatment of age-related macular degeneration with photodynamic therapy (TAP) Study Group. Arch Ophthalmol. 1999;117(10):1329–45.[PubMed]
- 50.Schmidt-Erfurth U, Hasan T. Mechanisms of action of photodynamic therapy with verteporfin for the treatment of age-related macular degeneration. Surv Ophthalmol. 2000;45(3):195–214. [PubMed]
- 51.Verteporfin therapy of subfoveal choroidal neovascularization in age-related macular degeneration: two-year results of a randomized clinical trial including lesions with occult with no classic choroidal neovascularization–verteporfin in photodynamic therapy report 2. Am J Ophthalmol. 2001;131(5):541–60. [PubMed]
- 52.Bressler NM, Arnold J, Benchaboune M, Blumenkranz MS, Fish GE, Gragoudas ES. et al. Verteporfin therapy of subfoveal choroidal neovascularization in patients with age-related macular degeneration: additional information regarding baseline lesion composition’s impact on vision outcomes-TAP report No. 3. Arch Ophthalmol. 2002;120(11):1443–54. [PubMed]
- 53.Stevenson MR, Hart PM, Chakravarthy U, Mackenzie G, Bird AC, Owens SL. et al. Visual functioning and quality of life in the SubFoveal Radiotherapy Study (SFRADS): SFRADS report 2. Br J Ophthalmol. 2005;89(8):1045–51. [PubMed]
- 54.Goverdhan SV, Gibbs FA, Lotery AJ. Radiotherapy for age-related macular degeneration: no more pilot studies please. Eye. 2005;19(11):1137–41. [PubMed]
- 55.Marcus DM, Peskin E, Maguire M, Weissgold D, Alexander J, Fine S. et al. The age-related macular degeneration radiotherapy trial (AMDRT): one year results from a pilot study. Am J Ophthalmol. 2004;138(5):818–28. [PubMed]
- 56.Gragoudas ES, Adamis AP, Cunningham ET, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med.2004;351(27):2805–16. [PubMed]
- 57.Heir J, Shapiro H, Singh A. Randomized, Controlled Phase III Study of Ranibizumab (Lucentis) for Minimally Classic or Occult Neovascular Age-Related Macular Degeneration: Two-Year Efficacy Results of the MARINA Study. 2006.
- 58.Rosenfeld PJ, Moshfeghi AA, Puliafito CA. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmic Surg Lasers Imaging. 2005;36(4):331–5. [PubMed]
- 59.Avery RL, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ. Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology. 2006 Mar;113(3):363-72 e5.
- 60.Vinores SA. Anecortave (Alcon Laboratories). IDrugs. 2005;8(4):327–34. [PubMed]
- 61.D’Amico DJ, Goldberg MF, Hudson H, Jerdan JA, Krueger DS, Luna SP, et al. Anecortave acetate as monotherapy for treatment of subfoveal neovascularization in age-related macular degeneration: twelve-month clinical outcomes. Ophthalmology. 2003 Dec;110(12):2372-83; discussin 84-5.
- 62.Spaide RF, Sorenson J, Maranan L. Photodynamic therapy with verteporfin combined with intravitreal injection of triamcinolone acetonide for choroidal neovascularization. Ophthalmology. 2005;112(2):301–4. [PubMed]
- 63.Quiram PA, Gonzales CR, Schwartz SD. Severe steroid-induced glaucoma following intravitreal injection of triamcinolone acetonide. Am J Ophthalmol.2006;141(3):580–2. [PubMed]
- 64.Jonas JB, Kreissig I, Spandau UH, Harder B. Infectious and noninfectious endophthalmitis after intravitreal high-dosage triamcinolone acetonide. Am J Ophthalmol. 2006;141(3):579–80. [PubMed]
- 65.Guymer RH, Chiu AW, Lim L, Baird PN. HMG CoA reductase inhibitors (statins): do they have a role in age-related macular degeneration? Surv Ophthalmol.2005;50(2):194–206. [PubMed]
- 66.Sarks S, Arnold J, Killingsworth M, Sarks J. Early drusen formation in the normal and aging eye and their relation to age-related maculopathy: a clinicopathological study. British Journal of Ophthalmology. 1999;83:358–68. [PubMed]
- 67.McConnell V, Silvestri G. Age-related macular degeneration. Ulster Med J. 2005;74(2):82–92. [PubMed]
- 68.Provis JM, Penfold PL, Cornish EE, Sandercoe TM, Madigan MC. Anatomy and development of the macula: specialisation and the vulnerability to macular degeneration. Clin Exp Optom. 2005;88(5):269–81. [PubMed]
- 69.Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol.2006;51(2):137–52. [PubMed]
- 70.Arnold J, Sarks S. Age related macular degeneration. Clin Evid. 2004;(11):819–34. [PubMed]
- 71.Hageman G, Mullins R. Molecular composition of drusen as related to substructural phenotype. Molecular Vision. 1999;5:28. [PubMed]
- 72.Sarraf D, Gin T, Yu F, Brannon A, Owens SL, Bird AC. Long-term drusen study. Retina. 1999;19(6):513–9. [PubMed]
- 73.Klein R, Klein B, Jensen S, Meuer S. The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology.1997;104:7–21. [PubMed]
- 74.Spraul C, Grossniklaus H. Characteristics of drusen and Bruch’s membrane in postmortem eyes with age-related macular degeneration. Archives of Ophthalmology. 1997;115:267–73. [PubMed]
- 75.Sarks JP, Sarks SH, Killingsworth MC. Evolution of soft drusen in age-related macular degeneration. Eye. 1994;8(Pt 3)(3):269-83.
- 76.Guymer R, Bird A. Bruch’s membrane, drusen, and age-related macular degeneration. In: Marmor M, Wolfensberger T, editors. The Retinal Pigment Epithelium. New York: Oxford University Press; 1998. p. 693-705.
- 77.Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122(4):598–614. [PubMed]
- 78.Sivaprasad S, Bailey TA, Chong VN. Bruch’s membrane and the vascular intima: is there a common basis for age-related changes and disease? Clin Experiment Ophthalmol. 2005;33(5):518–23. [PubMed]
- 79.Marshall J, Hussain A, Starita C, Moore D, Patmore A. The Retinal Pigment Epithelium. New York: Oxford University Press; 1998.
- 80.Guymer R, Luthert P, Bird A. Changes in Bruch’s membrane and related structures with age. Progress in Retinal & Eye Research. 1999;18(1):59–90. [PubMed]
- 81.Hogan M, Alvarado J. Studies on the human macula. Aging changes in Bruch’s membrane. Archives of Ophthalmology. 1967;77:410–20. [PubMed]
- 82.Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. British Journal of Ophthalmology. 1976;60(5):324–41. [PubMed]
- 83.Green WR, McDonnell PJ, Yeo JH. Pathologic features of senile macular degeneration. Ophthalmology. 1985;92(5):615–27. [PubMed]
- 84.Feeney-Burns L, Ellersieck M. Age-related changes in the ultrastructure of Bruch’s membrane. American Journal of Ophthalmology. 1985;100:686–97. [PubMed]
- 85.Grindle C, Marshall J. Ageing changes in Bruch’s membrane and their functional implications. Trans Ophthal Soc UK. 1978;98:172–5. [PubMed]
- 86.Pauleikhoff D, Sheraidah G, Marshall J, Bird A, Wessing A. Biochemical and histochemical analysis of age related lipid deposits in Bruch’s membrane.Ophthalmologe. 1994;91(6):730–4. [PubMed]
- 87.Handa JT, Verzijl N, Matsunaga H, Aotaki-Keen A, Lutty GA, te Koppele JM. et al. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Investigative Ophthalmology and Visual Science. 1999;40(3):775–9. [PubMed]
- 88.Curcio C, Millican C, Bailey T, Kruth H. Accumulation of cholesterol with age in human Bruch’s membrane. Investigative Ophthalmology & Visual Science.2001;42:265–74. [PubMed]
- 89.Kamei M, Hollyfield J. TIMP-3 in Bruch’s membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999;40:2367–75. [PubMed]
- 90.Newsome D, Huh W, Green W. Bruch’s membrane age-related changes vary by region. Current Eye Research. 1987;6:1211–21. [PubMed]
- 91.Ishibashi T, Murata T, Hangai M, Nagai R, Horiuchi S, Lopez P. et al. Advanced glycation end products in age-related macular degeneration. Arch Ophthalmol.1998;116:1629–32. [PubMed]
- 92.Hewitt A, Nakazawa K, Newsome D. Analysis of newly synthesized Bruch’s membrane proteoglycans. Investigative Ophthalmology and Visual Science.1989;30:478–86. [PubMed]
- 93.Karwatowski W, Jeffries T, Duance V, Albon J, Bailey A, Easty D. Preparation of Bruch’s membrane and analysis of the age-related changes in the structural collagens. Br J Ophthalmol. 1995;79(10):944–52. [PubMed]
- 94.Haimovici R, Gantz DL, Rumelt S, Freddo TF, Small DM. The lipid composition of drusen, Bruch’s membrane, and sclera by hot stage polarizing light microscopy.Invest Ophthalmol Vis Sci. 2001;42(7):1592–9. [PubMed]
- 95.Li CM, Chung BH, Presley JB, Malek G, Zhang X, Dashti N. et al. Lipoprotein-like particles and cholesteryl esters in human Bruch’s membrane: initial characterization. Invest Ophthalmol Vis Sci. 2005;46(7):2576–86. [PubMed]
- 96.Starita C, Hussain A, Pagliarini S, Marshall J. Hydrodynamics of ageing Bruch’s membrane: implications for macular disease. Experimental Eye Research.1996;62:565–72. [PubMed]
- 97.Moore D, Clover G. The effect of age on the macromolecuar permeability of human Bruch’s membrane. Invest Ophthalmol Vis Sci. 2001;42:2970–5. [PubMed]
- 98.Hussain A, Rowe L, Marshall J. Age-related alterations in the diffusional transport of amino acids across the human Bruch’s-choroid complex. Journal of the Optical Society of America. 2002;19:166–72. [PubMed]
- 99.Moore D, Hussain A, Marshall J. Age-related variation in the hydraulic conductivity of Bruch’s membrane. Invest Ophthalmol Vis Sci. 1995;36:1290–7. [PubMed]
- 100.Chong NH, Keonin J, Luthert PJ, Frennesson CI, Weingeist DM, Wolf RL. et al. Decreased thickness and integrity of the macular elastic layer of Bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am J Pathol. 2005;166(1):241–51. [PubMed]
- 101.Nackman G, Karkowski F, Halpern V, Gaetz H, Tilson M. Elastin degradation products induce adventitial angiogenesis in the Anidjar/Dobrin rat aneurysm model.Surgery. 1997;122:39–44. [PubMed]
- 103.Marshall J, Hussain A, Starita C, Moore D, Patmore A. Aging and Bruch’s membrane. In: Marmor M, Wolfensberger T, editors. The Retinal Pigment Epithelium. New York: Oxford University Press; 1998. p. 669-92.
- 104.Holz F, Sheraiadah G, Pauleikhoff D, Marshall J, Bird A. Analysis of lipid deposits extracted from human macular and peripheral Bruch’s membrane. Archives of Ophthalmology. 1994;112:402–6. [PubMed]
- 105.Pauleikhoff D, Harper C, Marshall J, Bird A. Aging changes in Bruch’s membrane. Ophthalmology. 1990;97:171–8. [PubMed]
- 106.Anderson D, Ozaki S, Nealon M, Neitz J, Mullins R, Hageman G. et al. Local cellular sources of apolipoprotein E in the human retina and retinal pigmented epithelium: Implications for the process of drusen formation. American J Ophthalmol. 2001;131:767–81.
- 107.Farkas T, Sylvester V, Archer D, Altona M. The histochemistry of drusen. American Journal of Ophthalmology. 1971;71:1206–15. [PubMed]
- 108.Green W, Enger C. Age-related macular degeneration histopathologic studies. Ophthalmology. 1993;100:1519–35. [PubMed]
- 109.Kliffen M, van der Schaft TL, Mooy CM, de Jong PT. Morphologic changes in age-related maculopathy. Microscopy Research & Technique. 1997;36(2):106–22.[PubMed]
- 110.Feher J, Valu L. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1967;173(2):162–7. [PubMed]
- 111.Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100(10):1519–35. [PubMed]
- 112.Loffler KU, Lee WR. Basal linear deposit in the human macula. Graefes Arch Clin Exp Ophthalmol. 1986;224(6):493–501. [PubMed]
- 113.van der Schaft TL, de Bruijn WC, Mooy CM, Ketelaars DA, de Jong PT. Is basal laminar deposit unique for age-related macular degeneration? Archives of Ophthalmology. 1991;109(3):420–5. [PubMed]
- 114.van der Schaft T, Mooy C, de Bruijn W, Oron F, Mulder P, de Jong P. Histological features of age-related macular degeneration. A statistical analysis.Ophthalmology. 1992;99:278–86. [PubMed]
- 115.Spraul CW, Lang GE, Grossniklaus HE. Morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37(13):2724–35. [PubMed]
- 116.Milam AH, Curcio CA, Cideciyan AV, Saxena S, John SK, Kruth HS. et al. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology. 2000;107(12):2256–66. [PubMed]
- 117.Kliffen M, Mooy CM, Luider TM, Huijmans JG, Kerkvliet S, de Jong PT. Identification of glycosaminoglycans in age-related macular deposits. Archives of Ophthalmology. 1996;114(8):1009–14. [PubMed]
- 118.Kliffen M, Mooy CM, Luider TM, de Jong PT. Analysis of carbohydrate structures in basal laminar deposit in aging human maculae. Investigative Ophthalmology and Visual Science. 1994;35(7):2901–5. [PubMed]
- 119.van der Schaft T, Mooy C, de Bruijn W, Bosman F, de Jong P. Immunohistochemical light and electron microscopy of basal laminar deposit. Graefe’s Archives for Clinical and Experimental Ophthalmology. 1994;232:40–6.
- 120.Knupp C, Amin S, Munro P, Luthert P, Squire J. Collagen VI assemblies in age-related macular degeneration. J Struct Biol. 2002;139:181–9. [PubMed]
- 121.Curcio CA, Presley JB, Millican CL, Medeiros NE. Basal deposits and drusen in eyes with age-related maculopathy: evidence for solid lipid particles. Exp Eye Res. 2005;80(6):761–75. [PubMed]
- 122.Sarks S, Van Driel D, Maxwell L, Killingsworth M. Softening of drusen and subretinal neovascularization. Transactions of the Ophthalmological Society of the United Kingdom. 1980;100:414–22.
- 123.Okubo A, Rosa R, Bunce C, Alexander R, Fan J, Bird A. et al. Relationship of age changes in retinal pigment epithelium and Bruch’s membrane. IOVS.1999;40:443–9.
- 124.Hogan M. Role of the retinal pigment epithelium in macular disease. Transactions of the American Academy of Ophthalmology and Otolaryngology. 1972;76:64–80.
- 125.Bird A. Pathophysiology of AMD. In: Hampton G, PT N, editors. Age-related macular degeneration: principles and practice. New York: Raven Press; 1992. p. chap. 3.
- 126.Dorey C, Wu G, Ebenstein D. Cell loss in the aging retina: Relationship to lipofuscin accumulation and macular degeneration. IOVS. 1989;30:1691–9.
- 127.Bird AC. Bruch’s membrane change with age. Br J Ophthalmol. 1992;76(3):166–8. [PubMed]
- 128.Katz ML, Robison WG. Age-related changes in the retinal pigment epithelium of pigmented rats. Exp Eye Res. 1984;38(2):137–51. [PubMed]
- 129.Feeney-Burns L, Hilderbrand E, Eldridge S. Aging human RPE: morphometric analysis of macular, equatorial and peripheral cells. IOVS. 1984;25:195–200.
- 130.Wing G, Blanchard G, Weiter J. Topography and age relationship of lipofuscin concentration in the RPE. IOVS. 1978;7:601–7.
- 131.Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J Biol Chem. 1995;270(32):18825–30. [PubMed]
- 132.Holz F, Bellman C, Staudt S, Schutt F, Volcker H. Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2001;42:1051–6. [PubMed]
- 133.Katz ML. Potential role of retinal pigment epithelial lipofuscin accumulation in age-related macular degeneration. Arch Gerontol Geriatr. 2002;34(3):359–70.[PubMed]
- 134.Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005;80(5):595–606. [PubMed]
- 135.Ramrattan R, van der Schaft T, Mooy C, de Bruijn W, Mulder P, de Jong P. Morphometric analysis of Bruch’s membrane, the choriocapillaris, and the choroid in aging. Investigative Ophthalmology & Visual Science. 1994;35:2857–64. [PubMed]
- 136.McLeod DS, Taomoto M, Otsuji T, Green WR, Sunness JS, Lutty GA. Quantifying changes in RPE and choroidal vasculature in eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2002;43(6):1986–93. [PubMed]
- 137.Midena E, Segato T, Blarzino MC, Degli Angeli C. Macular drusen and the sensitivity of the central visual field. Documenta Ophthalmologica. 1994;88(2):179–85.[PubMed]
- 138.Curcio C, Medeiros N, Millican C. Photoreceptor loss in age-related macular degeneration. Investigative Ophthalmology & Visual Science. 1996;37:1236–49.[PubMed]
- 139.Jackson GR, Owsley C. Visual dysfunction, neurodegenerative diseases, and aging. Neurol Clin. 2003;21(3):709–28. [PubMed]
- 140.Li J, Tso MO, Lam TT. Reduced amplitude and delayed latency in foveal response of multifocal electroretinogram in early age related macular degeneration. Br J Ophthalmol. 2001;85(3):287–90. [PubMed]
- 141.Owsley C, Jackson GR, Cideciyan AV, Huang Y, Fine SL, Ho AC. et al. Psychophysical evidence for rod vulnerability in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41(1):267–73. [PubMed]
- 142.Jackson GR, Owsley C, Curcio CA. Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res Rev. 2002;1(3):381–96.[PubMed]
- 143.Owsley C, Jackson GR, White M, Feist R, Edwards D. Delays in rod-mediated dark adaptation in early age-related maculopathy. Ophthalmology.2001;108(7):1196–202. [PubMed]
- 144.Jackson GR, McGwin G, Phillips JM, Klein R, Owsley C. Impact of aging and age-related maculopathy on activation of the a-wave of the rod-mediated electroretinogram. Invest Ophthalmol Vis Sci. 2004;45(9):3271–8. [PubMed]
- 145.Owsley C, McGwin G, Jackson GR, Heimburger DC, Piyathilake CJ, Klein R. et al. Effect of short-term, high-dose retinol on dark adaptation in aging and early age-related maculopathy. Invest Ophthalmol Vis Sci. 2006;47(4):1310–8. [PubMed]
- 146.Stangos N, Voutas S, Topouzis F, Karampatakis V. Contrast sensitivity evaluation in eyes predisposed to age-related macular degeneration and presenting normal visual acuity. Ophthalmologica. 1995;209:194–8. [PubMed]
- 147.Sunness J, Massof R, Johnson M, Bressler N, Bressler S, Fine S. Diminished foveal sensitivity may predict the development of advanced age-related macular degeneration. Ophthalmology. 1989;96:375–81. [PubMed]
- 148.Kleiner RC, Enger C, Alexander MF, Fine SL. Contrast sensitivity in age-related macular degeneration. Arch Ophthalmol. 1988;106(1):55–7. [PubMed]
- 149.Sandberg MA, Gaudio AR. Slow photostress recovery and disease severity in age-related macular degeneration. Retina. 1995;15(5):407–12. [PubMed]
- 150.Eisner A, Klein ML, Zilis JD, Watkins MD. Visual function and the subsequent development of exudative age-related macular degeneration. Invest Ophthalmol Vis Sci. 1992;33(11):3091–102. [PubMed]
- 151.Eisner A, Stoumbos VD, Klein ML, Fleming SA. Relations between fundus appearance and function. Eyes whose fellow eye has exudative age-related macular degeneration. Invest Ophthalmol Vis Sci. 1991;32(1):8–20. [PubMed]
- 152.Johnson PT, Brown MN, Pulliam BC, Anderson DH, Johnson LV. Synaptic pathology, altered gene expression, and degeneration in photoreceptors impacted by drusen. Invest Ophthalmol Vis Sci. 2005;46(12):4788–95. [PubMed]
- 153.Johnson PT, Lewis GP, Talaga KC, Brown MN, Kappel PJ, Fisher SK. et al. Drusen-associated degeneration in the retina. Invest Ophthalmol Vis Sci.2003;44(10):4481–8. [PubMed]
- 154.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune- mediated processes at the rpe-bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20(6):705–32. [PubMed]
- 155.Anderson D, Mullins R, Hageman G. LV J. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–31.[PubMed]
- 156.Killingsworth M, Sarks J, Sarks S. Macrophages related to Bruch’s membrane in age-related macular degeneration. Eye. 1990;4:613–21. [PubMed]
- 157.Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and Aetiological Aspects of Macular Degeneration. Progress in Retinal Research. London: Elsevier Science Ltd; 2001. p. 385-414.
- 158.Heriot W, Henkind P, Bellhorn R, Burns M. Choroidal neovascularization can digest Bruch’s membrane. A prior break is not essential. Ophthalmology.1984;91(12):1603–8. [PubMed]
- 159.Penfold P, Killingsworth M, Sarks S. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of Bruch’s membrane. Australian Journal of Ophthalmology. 1984;12:23–31. [PubMed]
- 160.Dastgheib K, Green W. Granulomatous reaction to Bruch’s membrane in age-related macular degeneration. Archives of Ophthalmology. 1994;112:813–8.[PubMed]
- 161.Hageman G, Mullins R, Russell S, Johnson L, Anderson D. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB Journal. 1999;13(3):477–84. [PubMed]
- 162.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. Faseb J. 2000;14(7):835–46. [PubMed]
- 163.Johnson L, Ozaki S, Staples M, Erickson P, Anderson D. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70:441–9.[PubMed]
- 164.Johnson L, Leitner W, Staples M, Anderson D. Complement activation and inflammatory processes in drusen formation and age related macular degeneration.Exp Eye Res. 2001;73:887–96. [PubMed]
- 165.Mullins RF, Johnson LV, Anderson DH, Hageman GS. Characterization of drusen-associated glycoconjugates. Ophthalmology. 1997;104(2):288–94. [PubMed]
- 166.Russell SR, Mullins RF, Schneider BL, Hageman GS. Location, substructure, and composition of basal laminar drusen compared with drusen associated with aging and age-related macular degeneration. Am J Ophthalmol. 2000;129(2):205–14. [PubMed]
- 167.Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye. 2001;15(Pt 3):390–5. [PubMed]
- 168.Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99(18):11830–5. [PubMed]
- 169.Malek G, Li CM, Guidry C, Medeiros NE, Curcio CA. Apolipoprotein B in cholesterol-containing drusen and basal deposits of human eyes with age-related maculopathy. Am J Pathol. 2003;162(2):413–25. [PubMed]
- 170.Anderson D, Talaga K, Rivest A, Barron E, Hageman G, Johnson L. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78:243–56. [PubMed]
- 171.Peto T, Lengyel I, Frederickson C, Vankuijk F. Zinc in Drusen. ARVO Abstract. 2005 May 3, 2005.
- 190.Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41(4):355–67. [PubMed]
- 191.Pangburn MK, Pangburn KL, Koistinen V, Meri S, Sharma AK. Molecular mechanisms of target recognition in an innate immune system: interactions among factor H, C3b, and target in the alternative pathway of human complement. J Immunol. 2000;164(9):4742–51. [PubMed]
- 192.Zipfel PF. Complement factor H: physiology and pathophysiology. Semin Thromb Hemost. 2001;27(3):191–9. [PubMed]
- 193.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI. et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102(20):7227–32. [PubMed]
- 194.Johnson LV, Talaga KC, Rivest AJ, Staples MK, Radeke MJ, Barron E, et al. Characterization of Beta Amyloid-Containing Vesicles within Drusen. ARVO Abstract. 2003.
- 195.Hageman G, Luthert P, Chong N, Johnson L, Anderson D, Mullins R. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–32. [PubMed]
- 196.Miller DM, Espinosa-Heidmann DG, Legra J, Dubovy SR, Suner IJ, Sedmak DD. et al. The association of prior cytomegalovirus infection with neovascular age-related macular degeneration. Am J Ophthalmol. 2004;138(3):323–8. [PubMed]
- 197.Klein M, Mauldin W, Stoumbos V. Heredity and age-related macular degeneration. Observations in monozygotic twins. Archives of Ophthalmology.1994;112(7):932–7. [PubMed]
- 198.Heiba I, Elston R, Klein B, Klein R. Sibling correlations and segregation analysis of age-related maculopathy: the Beaver Dam Eye Study. Genetic Epidemiology.1994;11:51–67. [PubMed]
- 199.Seddon J, Ajani U, Mitchell B. Familial aggregation of age-related maculopathy. American Journal of Ophthalmology. 1997;123:199–206. [PubMed]
- 200.Meyers SM, Greene T, Gutman FA. A twin study of age-related macular degeneration. Am J Ophthalmol. 1995;120(6):757–66. [PubMed]
- 201.Hammond CJ, Webster AR, Snieder H, Bird AC, Gilbert CE, Spector TD. Genetic influence on early age-related maculopathy: a twin study. Ophthalmology.2002;109(4):730–6. [PubMed]
- 202.Abecasis GR, Yashar BM, Zhao Y, Ghiasvand NM, Zareparsi S, Branham KE. et al. Age-related macular degeneration: a high-resolution genome scan for susceptibility loci in a population enriched for late-stage disease. Am J Hum Genet. 2004;74(3):482–94. [PubMed]
- 203.Iyengar SK, Song D, Klein BE, Klein R, Schick JH, Humphrey J. et al. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am J Hum Genet. 2004;74(1):20–39. [PubMed]
- 204.Klein ML, Schultz DW, Edwards A, Matise TC, Rust K, Berselli CB. et al. Age-related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Archives of Ophthalmology. 1998;116(8):1082–8. [PubMed]
- 205.Majewski J, Schultz DW, Weleber RG, Schain MB, Edwards AO, Matise TC. et al. Age-related macular degeneration–a genome scan in extended families. Am J Hum Genet. 2003;73(3):540–50. [PubMed]
- 206.Schick JH, Iyengar SK, Klein BE, Klein R, Reading K, Liptak R. et al. A whole-genome screen of a quantitative trait of age-related maculopathy in sibships from the Beaver Dam Eye Study. Am J Hum Genet. 2003;72(6):1412–24. [PubMed]
- 207.Seddon JM, Santangelo SL, Book K, Chong S, Cote J. A genomewide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am J Hum Genet. 2003;73(4):780–90. [PubMed]
- 208.Weeks DE, Conley YP, Mah TS, Paul TO, Morse L, Ngo-Chang J. et al. A full genome scan for age-related maculopathy. Hum Mol Genet. 2000;9(9):1329–49.[PubMed]
- 209.Weeks DE, Conley YP, Tsai HJ, Mah TS, Rosenfeld PJ, Paul TO. et al. Age-related maculopathy: an expanded genome-wide scan with evidence of susceptibility loci within the 1q31 and 17q25 regions. Am J Ophthalmol. 2001;132(5):682–92. [PubMed]
- 210.Weeks DE, Conley YP, Tsai HJ, Mah TS, Schmidt S, Postel EA. et al. Age-Related Maculopathy: A Genomewide Scan with Continued Evidence of Susceptibility Loci within the 1q31, 10q26, and 17q25 Regions. Am J Hum Genet. 2004;75(2)
- 211.Fisher SA, Abecasis GR, Yashar BM, Zareparsi S, Swaroop A, Iyengar SK. et al. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet. 2005;14(15):2257–64. [PubMed]
- 212.Schultz D, Klein M, Humpert A, Luzier C, Persun V, Schain M, et al. Analysis of the ARMD1 locus: Evidence that a mutation in HEMICENTIN-1 is associated with age-related macular degeneration in a large family. Hum Mol Genet. 2003.
- 213.Conley YP, Thalamuthu A, Jakobsdottir J, Weeks DE, Mah T, Ferrell RE. et al. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age-related maculopathy. Hum Mol Genet. 2005;14(14):1991–2002. [PubMed]
- 214.Haines JL, Schnetz-Boutaud N, Schmidt S, Scott WK, Agarwal A, Postel EA. et al. Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest Ophthalmol Vis Sci. 2006;47(1):329–35. [PubMed]
- 215.Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA. et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351(4):346–53. [PubMed]
- 216.Zareparsi S, Buraczynska M, Branham KE, Shah S, Eng D, Li M. et al. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Hum Mol Genet. 2005;14(11):1449–55. [PubMed]
- 217.Souied EH, Benlian P, Amouyel P, Feingold J, Lagarde JP, Munnich A. et al. The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. American Journal of Ophthalmology. 1998;125(3):353–9. [PubMed]
- 218.Klaver CC, Kliffen M, van Duijn CM, Hofman A, Cruts M, Grobbee DE. et al. Genetic association of apolipoprotein E with age-related macular degeneration.American Journal of Human Genetics. 1998;63(1):200–6. [PubMed]
- 219.Baird PN, Guida E, Chu DT, Vu HT, Guymer RH. The epsilon2 and epsilon4 alleles of the apolipoprotein gene are associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45(5):1311–5. [PubMed]
- 220.Allikmets R, Shroyer N, Singh N, Seddon J, Lewis R, Bernstein P. et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration.Science. 1997;277:1805–7. [PubMed]
- 221.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C. et al. Complement factor H polymorphism in age-related macular degeneration. Science.2005;308(5720):385–9. [PubMed]
- 222.Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science.2005;308(5720):421–4. [PubMed]
- 223.Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P. et al. Complement factor H variant increases the risk of age-related macular degeneration.Science. 2005;308(5720):419–21. [PubMed]
- 224.Zareparsi S, Branham KE, Li M, Shah S, Klein RJ, Ott J. et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet. 2005;77(1):149–53. [PubMed]
- 225.Magnusson KP, Duan S, Sigurdsson H, Petursson H, Yang Z, Zhao Y. et al. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD.PLoS Med. 2006;3(1):e5. [PubMed]
- 226.Sepp T, Khan JC, Thurlby DA, Shahid H, Clayton DG, Moore AT. et al. Complement factor H variant Y402H is a major risk determinant for geographic atrophy and choroidal neovascularization in smokers and nonsmokers. Invest Ophthalmol Vis Sci. 2006;47(2):536–40. [PubMed]
- 227.Souied EH, Leveziel N, Richard F, Dragon-Durey MA, Coscas G, Soubrane G. et al. Y402H complement factor H polymorphism associated with exudative age-related macular degeneration in the French population. Mol Vis. 2005;11:1135–40. [PubMed]
- 228.Okamoto H, Umeda S, Obazawa M, Minami M, Noda T, Mizota A. et al. Complement factor H polymorphisms in Japanese population with age-related macular degeneration. Mol Vis. 2006;12:156–8. [PubMed]
- 229.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K. et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38(4):458–62. [PubMed]
- 230.Lokki ML, Koskimies SA. Allelic differences in hemolytic activity and protein concentration of BF molecules are found in association with particular HLA haplotypes. Immunogenetics. 1991;34(4):242–6. [PubMed]
- 231.Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005;77(3):389–407. [PubMed]
- 232.Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T. et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14(21):3227–36. [PubMed]
- 233.Schmidt S, Hauser MA, Scott WK, Postel EA, Agarwal A, Gallins P. et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006;78(5):852–64. [PubMed]
- 234.Saunders RE, Goodship TH, Zipfel PF, Perkins SJ. An interactive web database of factor H-associated hemolytic uremic syndrome mutations: insights into the structural consequences of disease-associated mutations. Hum Mutat. 2006;27(1):21–30. [PubMed]
- 235.Heinen S, Sanchez-Corral P, Jackson MS, Strain L, Goodship JA, Kemp EJ. et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat. 2006;27(3):292–3. [PubMed]
- 236.Warwicker P, Goodship TH, Donne RL, Pirson Y, Nicholls A, Ward RM. et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int.1998;53(4):836–44. [PubMed]
- 237.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, Lopez-Trascasa M, Sanchez-Corral P. et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet.2005;14(5):703–12. [PubMed]
- 238.Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G. et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12(24):3385–95. [PubMed]
- 239.Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006 Apr 18.
- 240.Atkinson JP, Liszewski MK, Richards A, Kavanagh D, Moulton EA. Hemolytic uremic syndrome: an example of insufficient complement regulation on self-tissue.Ann N Y Acad Sci. 2005;1056:144–52. [PubMed]
- 241.Abrera-Abeleda MA, Nishimura C, Smith JL, Sethi S, McRae JL, Murphy BF, et al. Variations in the Complement Regulatory Genes Factor H (CFH) and Factor H Related 5 (CFHR5) are Associated with Membranoproliferative Glomerulonephritis Type II (Dense Deposit Disease). J Med Genet. 2005 Nov 18.
- 242.Appel GB, Cook HT, Hageman G, Jennette JC, Kashgarian M, Kirschfink M. et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol. 2005;16(5):1392–403. [PubMed]
- 243.Robman L, Mahdi O, McCarty C, Dimitrov P, Tikellis G, McNeil J. et al. Exposure to Chlamydia pneumoniae infection and progression of age-related macular degeneration. Am J Epidemiol. 2005;161(11):1013–9. [PubMed]
- 244.Erkkila H, Raitta C, Niemi K. Ocular findings in four siblings with pseudoxanthoma elasticum. Acta Ophthalmol (Copenh). 1983;61:589–99. [PubMed]
- 245.Zee RY, Diehl KA, Ridker PM. Complement factor H Y402H gene polymorphism, C-reactive protein, and risk of incident myocardial infarction, ischaemic stroke, and venous thromboembolism: A nested case-control study. Atherosclerosis. 2005 Oct 13.
- 246.Zee RY, Diehl KA, Ridker PM. Complement factor H Y402H gene polymorphism, C-reactive protein, and risk of incident myocardial infarction, ischaemic stroke, and venous thromboembolism: A nested case-control study. Atherosclerosis. 2006;187(2):332–5. [PubMed]
- 247.Fung AE, Lalwani GA, Rosenfeld PJ, Dubovy SR, Michels S, Feur WJ, Puliafito CA, Davis JL, Flynn HW, Esquiabro M. An optical coherence tomography-guided, variable dosing regimen with intravitreal ranibizumab (Lucentis) for neovascular, age-related macular degeberation. Am J Ophthalmol. 2007;143(4):566–83.[PubMed]
- 248.Kaiser PK. Verteporfin photodynamic therapy and anti-angiogenic drugs: potential for combination therapy in exudative age-related macular degeneraion. Curr med res Opin. 2007;23(3):477–87. [PubMed]
- 249.VEGF Inhibition Study in Ocular Neovascularization (V.I.S.I.O.N.) Clinical Trial Group, D’Amico DJ, Masonson HN, Patel M, Adamis AP, Cunningham ET Jr, Guyer DR, Katz B. Pegaptanib sodium for neovascular age-related macular degeneration: two-year safety results of the two prospective, multicenter, controlled clinical trials. Ophthalmology. 2006 Jun;113(6):992-1001.e6. Epub 2006 Apr 27.
- 250.Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S. ANCHOR Study Group.; Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–44. [PubMed]
- 251.Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY. MARINA Study Group.; Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–3. [PubMed]
| The author Gregory S. Hageman, Ph.D., is a John A. Moran Presidential Professor of Ophthalmology and Visual Sciences, and Director of the Moran Center for Translational Medicine at the University of Utah. He is a graduate of the University of Southern California, where he conducted both his undergraduate and graduate studies in biology and marine biology. Most of his previous research career was at the Iowa Carver College of Medicine and his primary research interests have always been assessment of pathways involved in the etiology of age-related macular degeneration (AMD). Dr. Hageman is an active member of various professional and honorary organizations including the Macula Society and has also served on a number of national and international advisory boards, service panels and review committee. Moreover, he serves or has served as an advisor to Genentech, Alcon, Pfizer, OccuLogix, Novartis, Tanox and American Home Products. Dr. Hageman is an author or co-author of more than 100 refereed and invited publications, as well as multiple issued patents and pending patent applications. He was recently invited to present his research findings to Congress. He received the Trustee Award from Foundation Fighting Blindness, the Roger Johnson Prize for Macular Degeneration. |  |